
基于全基因组重测序分析新疆细毛羊遗传多样性
2023-06-20 05:13陈开旭郭翠洁任斐儿李晓斌刘武军
新疆农业科学 2023年5期
陈开旭,郭翠洁,杨 帆,任斐儿,李晓斌,刘武军
(新疆农业大学动物科学学院,乌鲁木齐 830052)
0 引 言
【研究意义】我国羊毛生产主要集中在新疆、内蒙古、甘肃、青海、吉林等省(自治区),为我国目前细毛羊的主产区,占全国细毛羊总产量的97%[1],对畜牧业经济发展有重要影响[2]。新疆细毛羊是毛、肉兼用细毛羊品种,由高加索细毛羊公羊与哈萨克母羊、泊列考斯公羊与蒙古羊母羊进行杂交培育而成[3]。该品种适于干燥寒冷高原地区饲养,采食性好,生活力强,耐粗饲料等,利用全基因组重测序技术分析新疆细毛羊的遗传多样性,对发掘新疆细毛羊的遗传资源,保护其多样性基因库有重要意义。【前人研究进展】新疆绵羊品种较多[4],新疆细毛羊具有适应性强、毛质好、肉质多、产毛量高、净毛率高、体大、耐粗饲、遗传性能稳定等优良品质。开发和利用新疆细毛羊这一宝贵遗传资源,对我国畜牧业的发展具有重要意义[5]。遗传多样性的本质是地球上物种发生、进化和变异的基础[6,7],遗传多样性的研究方法已从传统的形态标记、染色体标记及生化标记等发展到分子水平遗传标记的研究[8]。【本研究切入点】目前有关基于全基因组重测序分析新疆细毛羊遗传多样性的研究文献较少,需利用新疆细毛羊的全基因组重测序数据,检测不同绵羊品种的核苷酸多态性和单倍型多态性。【拟解决的关键问题】通过全基因组重测序的方法寻找新疆细毛羊的高密度 SNPs 遗传标记,运用杂合度、连续纯合子区域、连锁不平衡分析评估新疆细毛羊的遗传多样性。
1 材料与方法
1.1 材 料
1.1.1 细毛羊
从伊犁巩乃斯种羊场随机选取10只新疆细毛羊(Xinjiang Fine Wool Sheep,XFW),通过颈静脉采血法采集10mL颈静脉血于含有抗凝剂(乙二胺四乙酸(EDTA)盐)的真空采血管中,置于-80℃冰箱中冻存备用。10只巴音布鲁克羊(Bayinbuluke Sheep,BYK)、10只策勒黑羊(Cele Black Sheep,CLE)、10只阿勒泰羊(Altay Sheep,ALT)的基因组数据下载自 NCBI 数据库(GenBank检索号SRP363313)的全基因组重测序数据。
1.1.2 DNA 提取与质量检测
按照血液全基因组DNA提取试剂盒(QIAGEN)操作步骤,从新疆细毛羊全血样品中提取基因组DNA。DNA样品的浓度和纯度(DNA样品中蛋白质和RNA等污染),通过NanoDrop 1000超微量紫外分光光度计进行检测,通过凝胶电泳试验判断,DNA样品的完整性和降解,综合评判DNA样品的质量。
1.1.3 全基因组重测序与序列
1.1.3.1 测序文库构建
全基因组重测序对基因组DNA的质量要求标准为:基因组DNA经琼脂糖凝胶电泳检测结果显示:条带单一且清晰明亮,无拖尾(降解)现象,基因组DNA经浓度测定结果显示:浓度≥ 50ng/μL,总量大于3 μg,OD260/OD280=1.8~2.0(无蛋白及RNA污染),则认为该DNA样品质量符合测序要求。
测序文库构建:(1)用超声波将检测合格的基因组DNA样品随机打断成长度约为500 bp的片段;(2)用末端修复酶修复DNA,以提高DNA连接入载体的效率;(3)片段3’ 末端连接多个碱基A尾;(4)连接测序接头;(5)对DNA片段进行选择;(6)PCR扩增DNA片段;(7)再次进行DNA片段的选择和纯化。
文库构建完成后使用荧光计(Qutit2.0)进行初定量,根据定量结果将DNA片段浓度稀释至1 ng/μL。使用Agilent 2100 bioanalyzer对Insert Size检测,确保Insert Size符合文库要求。使用Q-PCR方法准确定量文库的有效浓度。利用Illumina Hiseq 4000高通量测序平台对文库进行双末端全基因组重测序。
1.1.3.2 测序质量评估与过滤
对完成全基因组重测序后的基因组序列数据进行质量评估与过滤,包括:去除无法配对的读长(reads)和低质量的reads、去除reads两端的barcoding序列 和index序列。数据清理后,获得10个文库的20个.fastq文件。采用FastQC 软件对测序后的基因组序列质量进行统计,内容包括:绵羊基因组测序序列reads中C、T、G、A四种碱基的含量和偏好性;基因组序列中的reads数目和长度的总体分布;reads中每个碱基的总体质量。
1.1.3.3 全基因组序列比对及比对文件预处理
下载绵羊参考基因组序列文件(Oar_4.0)(https://www.ncbi.nlm.nih.gov/assembly/GCF_002742125.1)并建立其索引文件。绵羊参考基因组作为比对模板,使用 BWA MEM[9]程序对每个绵羊个体基因组文库测序生成的2个.fastq文件分别进行比对,生成2个.sam文件。10个DNA文库,共生成20个.sam文件。将下载的10只策勒黑羊、10只巴音布鲁克羊、10只阿勒泰羊个体的测序文件按照同样的方法比对,生成60个.sam 文件。整理和统计比对后的reads数量、比例、reads的配对率等结果。
1.1.3.4 遗传变异的鉴定、过滤和注释
依据与参考基因组比对得到的结果,使用SAMtools[10]和GATK(版本号:3.6-0-g89b7209)[11]两款软件包分别对个体单核苷酸多态性(SNP)进行检测,并相互确认检测结果。通过SAMtools软件的“mpileup”程序进行序列校准,将覆盖范围在4~200的变异筛选出来进行后续分析。通过GATK软件采用单体型检测方法对每只绵羊的基因组变异进行检测。采用过滤条件(MAF<0.05,丢失基因型>10%)过滤掉绵羊群体中具有较小等位基因频率(MAF)的SNP,得到高可信度的SNP。
使用GATK软件包对40个绵羊个体的基因组DNA序列进行插入或缺失(Indel)检测,保留1-30 bp的Indel以备后续分析。
下载绵羊的SNP数据(https://www.ncbi.nlm.nih.gov/snp/),并与研究中检测出的 SNP 进行比对验证,确认上述过程检测到的SNPs的准确性和可靠性。利用ANNOVAR软件[12,13]注释过滤和检验后的SNPs。
1.2 遗传多样性
使用 GATK(版本号:3.6-0-g89b7209)[11]软件对4个绵羊品种基因组中的变异位点和基因型进行检测,评估新疆细毛羊的遗传多样性。
1.2.1 杂合度(Heterozygosity,H)
采用mlRho程序[14]计算不同绵羊群体中每个个体的杂合度,作为评估不同绵羊品系群体间遗传多样性的指标之一。
1.2.2 连续纯合子区域
使用PLINK软件计算每个个体的连续性纯合片段长度和数量,设置参数[15-17]:chr-set 26 -maf 0.05 -homozyg-window-snp 50 -homozyg-snp 50 -homo-zyg-kb 300 -homozyg-density 50 -homozyg-gap 1000 -homozyg-window-missing 5 -homozyg-window-threshold 0.05 -homozyg-window-het 03.基于 ROH 的物理长度,将 ROH 片段按照 <0.5 Mb、0.5~1 Mb、1~2 Mb、2~4 Mb、>4 Mb 进行分类统计,计算不同分类区间的ROH数目的比例。基于ROH计算基因组近交系数FROH,基因组近交系数的计算公式如下:
式中,∑LROH为常染色体上ROH片段的长度之和,Lgenome为常染色体的物理长度之和。
1.2.3 连锁不平衡
使用Haploview软件计算不同群体成对r2值,用以评估群体的连锁不平衡程度,运用R软件包绘制不同绵羊品种间连锁不平衡衰减图。
2 结果与分析
2.1 基因组DNA提取质量检测
研究表明,DNA条带单一且无拖尾,DNA样品完整性较好、无严重降解。图1
注:M 泳道为λ-HindⅢ DNA Marker,1-10泳道为DNA样品
经试剂盒提取的基因组DNA OD260/OD280在1.8~2.0、基因组DNA浓度>50 ng/μL、基因组DNA总量>3 μg,基因组DNA的纯度和浓度均符合测序要求。表1
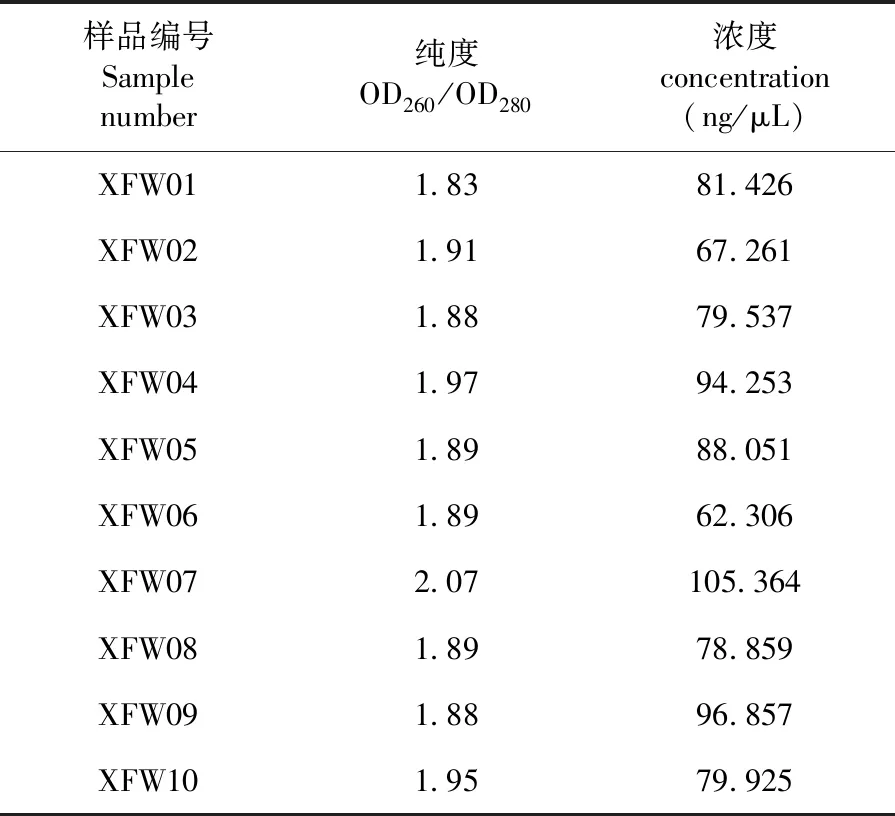
表1 新疆细毛羊DNA样品浓度及纯度检测
2.2 全基因组测序数据量和质量统计
研究表明,得到的测序总深度高达76.84×,个体的平均测序深度为 7.68×,平均的比对率为96.38%,平均的基因组覆盖度为97.88%,Q20(测序质量值≥20的碱基所占百分比)为96.97%,Q30(测序质量值≥30的碱基所占百分比)为91.93%。新疆细毛羊全基因组测序数据的比对率、全基因组覆盖度、Q20、Q30等反映测序质量的数据值均较高,测序质量较好,测序数据能够很好地反应个体的基因组信息。表2
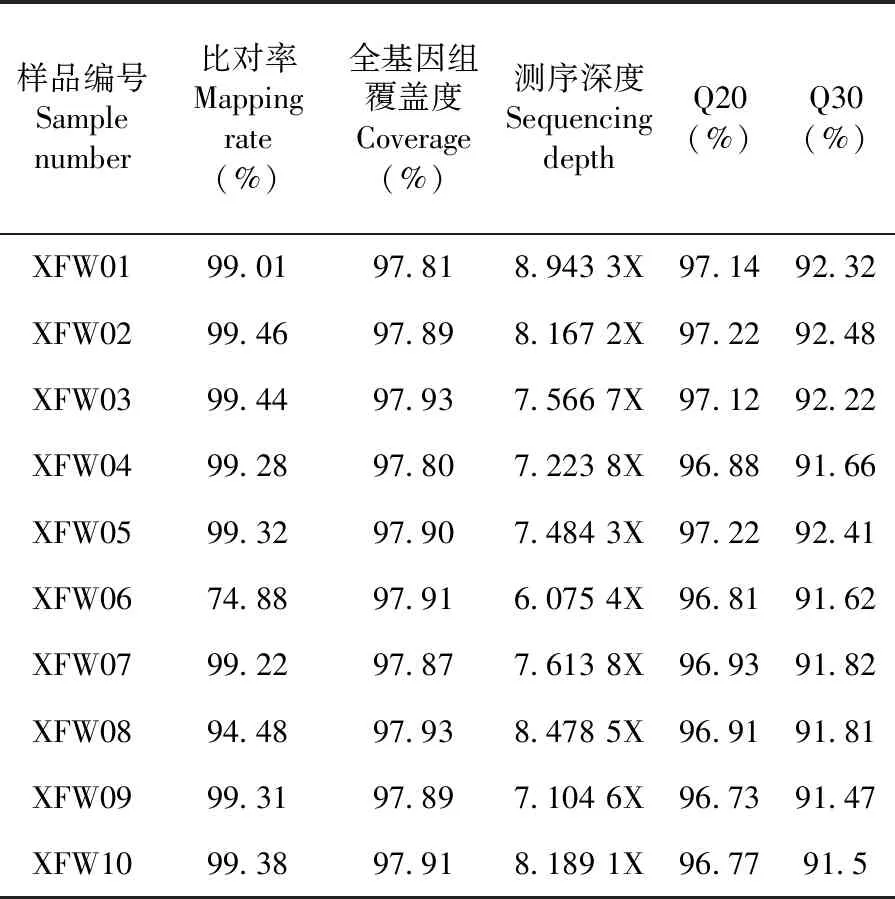
表2 全基因组测序数据质量统计
2.3 遗传变异的鉴定、过滤和注释
研究表明,最终获得了97 647 435个高质量的常染色SNP位点和15 886 270个Indel;其中,34 744 752个SNP位点(35.58%)和5 588 596个Indel(35.18%)位于内含子区域。还发现了97 986个错义变体和196 521个同义变体,以及1 394个缺失和981个插入导致的移码突变。表3
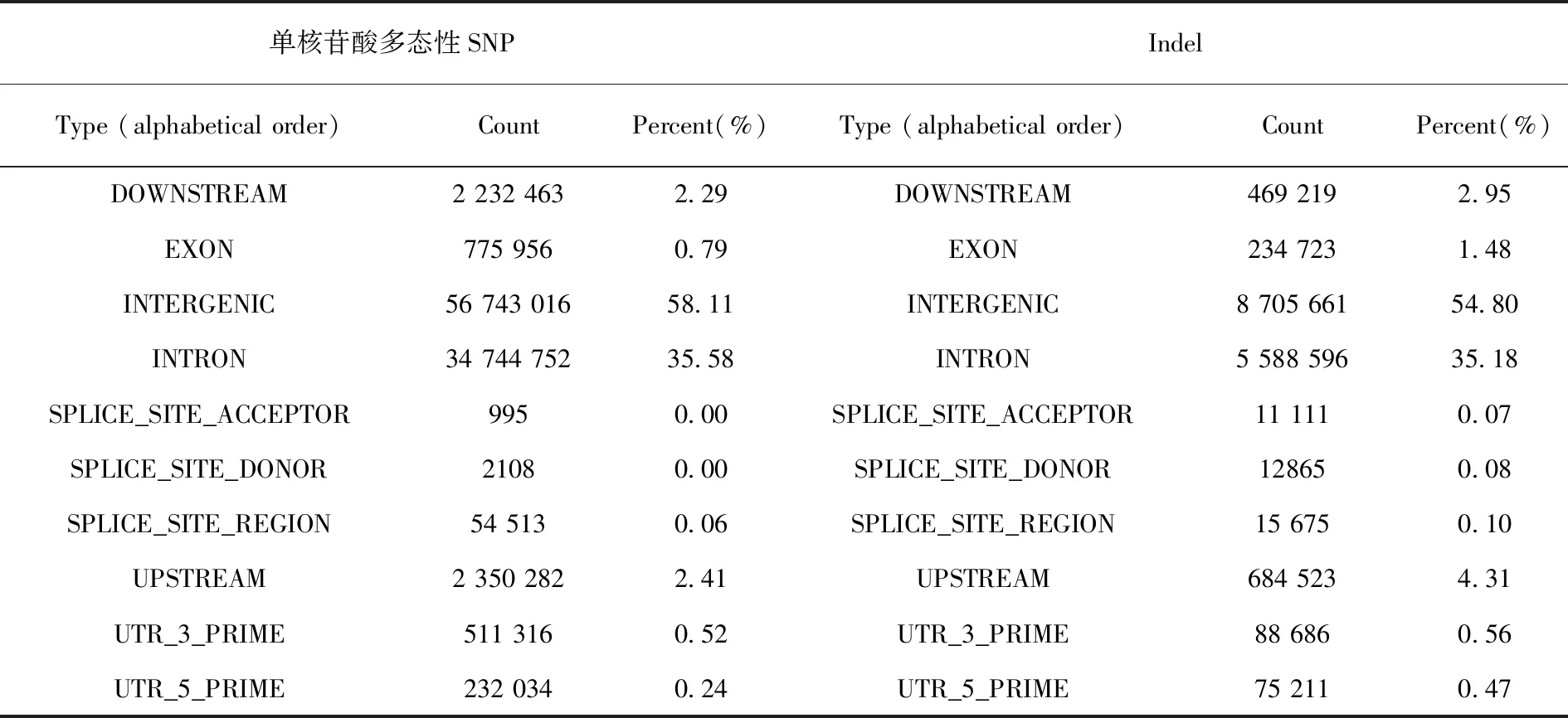
表3 新疆细毛羊全基因组重测序数据遗传变异鉴定、过滤和注释
2.4 4个绵羊群体的杂合度
研究表明,杂合度(Ho)和期望杂合度(He)的范围分别为0.175~0.233和0.239~0.245。在所有群体中,期望杂合度略高于观测杂合度,绵羊群体均有不同程度的近交或受到不同程度(人工或自然)选择的影响。新疆细毛羊的平均观测杂合度(Ho=0.196)极显著低于阿勒泰羊(Ho=0.222)(P<0.001),低于巴音布鲁克羊(Ho=0.201),略高于策勒黑羊(Ho=0.194)。图2
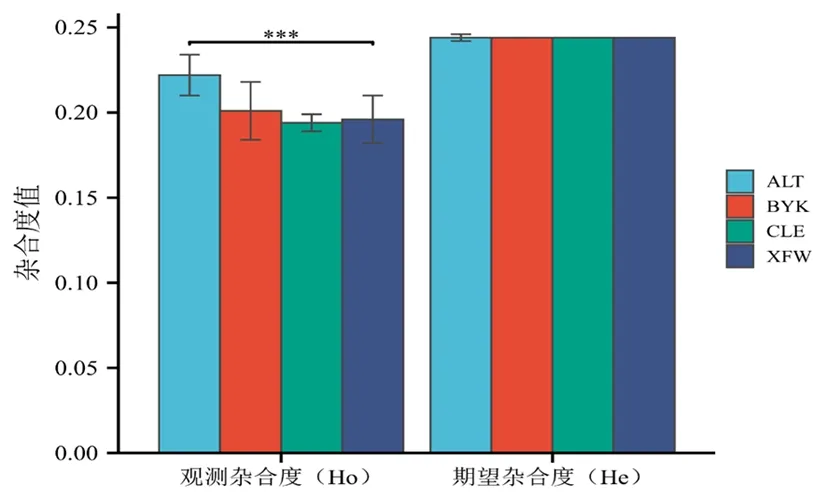
注:*表示不同绵羊群体间的杂合度具有显著差异(P<0.05),**表示不同绵羊群体间的杂合度具有极显著差异(P<0.01)
2.5 连续纯合子区域差异
研究表明,4个绵羊群体间的ROH长度区间差异较大,在47.985~178.833 Mb,而新疆细毛羊、策勒黑羊、巴音布鲁克羊、阿勒泰羊的平均ROH长度分别约为110.665、93.532、88.417和74.445 Mb,基因组近交系数分别约为0.042 3、0.035 8、0.033 8、0.028 5。表4
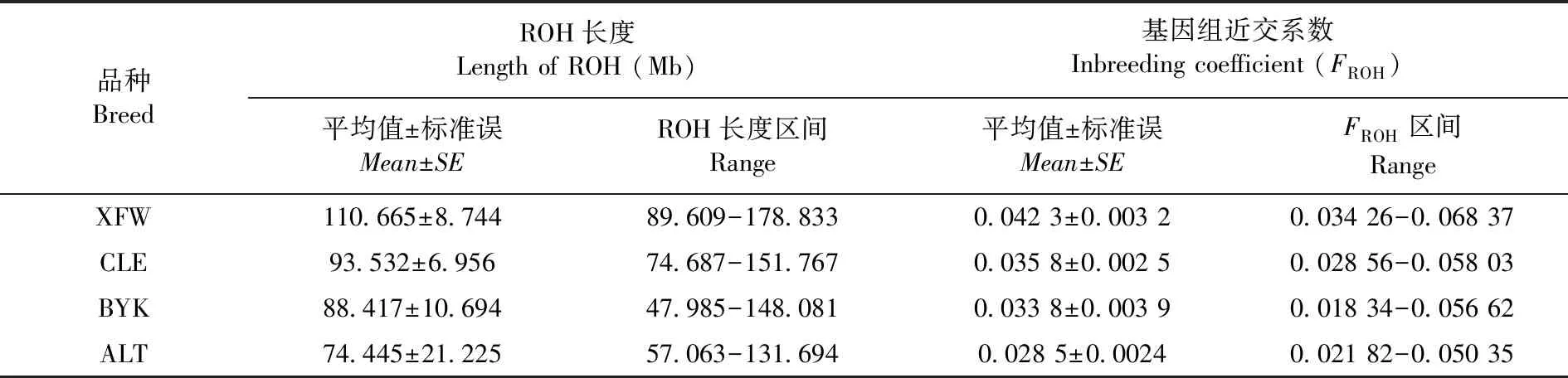
表4 4个绵羊群体的平均ROH长度和基因组近交系数
新疆细毛羊在<0.5 Mb范围的ROH总长度显著高于巴音布鲁克羊、策勒黑羊和阿勒泰羊(P<0.05),新疆细毛羊遗传多样性水平低于巴音布鲁克羊、策勒黑羊和阿勒泰羊。新疆细毛羊的平均ROH片段数量(215.8)显著高于策勒黑羊(166.2)、巴音布鲁克羊(152.9)和阿勒泰羊(150.2)(P<0.05),4个绵羊群体遗传多样性顺序:阿勒泰羊> 巴音布鲁克羊>策勒黑羊>新疆细毛羊。图3,图4
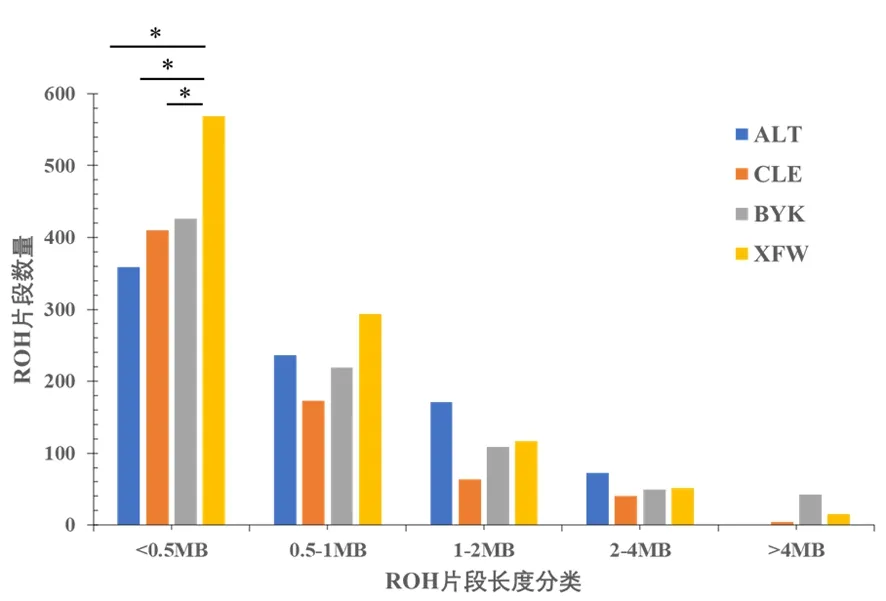
注:*表示不同绵羊群体间的ROH片段长度差异显著(P<0.05)
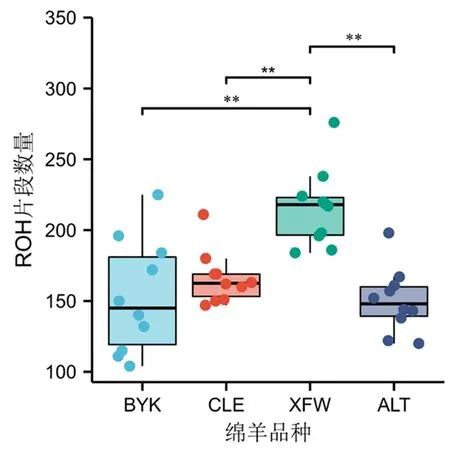
注:**表示不同绵羊群体间的平均ROH片段数量差异极显著(P<0.01)
2.6 4个绵羊群体的连锁不平衡性
研究表明,r2为0时,完全连锁平衡,群体独立遗传;r2等于1时,表示完全连锁不平衡。不同群体的绵羊整体连锁不平衡程度都较低,其中,新疆细毛羊的连锁不平衡程度相对最低,阿勒泰羊的连锁程度相对最高的,其次是巴音布鲁克羊和策勒黑羊,阿勒泰的遗传多样性最高,其次是巴音布鲁克羊,新疆细毛羊的遗传多样性最低。图5
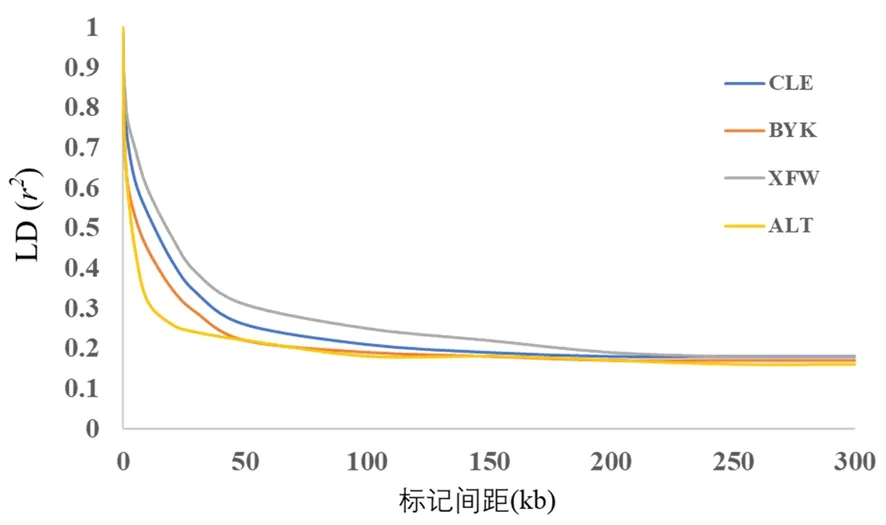
图5 4个绵羊群体的连锁不平衡变化
3 讨 论
3.1 新疆细毛羊的全基因组重测序和遗传变异检测
单核苷酸多态性(Single Nucleotide Polymorphisms,SNP) 指在基因组上单个核苷酸的变异(包括置换、颠换、缺失和插入),形成遗传标记,具有数量多,分布广泛,多态性丰富,易于快速、规模化筛查,便于基因分型等特点,其作为新的遗传标记对基因定位及相关疾病研究的意义亦非常重大。研究表明,部分基因的SNP位点与个体的发育性状有关,可以将此类基因作为选育生长发育性状的分子遗传标记[18,19],有利于加速 SNP 分子标记技术在绵羊育种、遗传分析中的应用[20]。研究采用全基因组重测序技术对10个新疆细毛羊个体进行全基因组重测序,通过遗传变异鉴定和基因型信息整合,对变异位点质量值,测序深度,有效信息比例等一系列的质量过滤和控制条件,最终获得了97 647 435个高质量的常染色SNP位点和15 886 270个Indel;其中,34 744 752个SNP位点(35.58%)和5 588 596个Indel(35.18%)位于内含子区域。此外,还发现了97 986个错义变体和196 521个同义变体,以及1 394个缺失和981个插入导致的移码突变。
3.2 新疆细毛羊的遗传多样性
3.2.1 杂合度
杂合度(Heterozygosity,H)是度量自然群体遗传变异的首选指标,表示在一个群体中某位点为杂合子的概率。群体杂合度能反映群体的遗传结构甚至是变化历史,其值介于0到1。当某一群体的期望杂合度(He)高于其观测杂合度(Ho)时,则群体被认为可能受到了选择或者近交影响;当某一群体的期望杂合度(He)低于其观测杂合度(Ho)时,则群体被认为可能引进了其他品种的血缘。杂合度越高意味着群体遗传多样性越丰富,反之,杂合度低群体遗传多样性低。新疆细毛羊的平均观测杂合度(0.196)极显著低于阿勒泰羊(0.222)(P<0.001),低于巴音布鲁克羊(0.201),略高于策勒黑羊(0.194),新疆细毛羊的遗传多样性水平显著低于阿勒泰羊,低于巴音布鲁克羊,略高于策勒黑羊。
3.2.2 连续纯合子区域分析
长纯合片段(rus of homozygosity,ROH)是一类基因组中出现的连续不间断的纯合现象,表现为一段染色体区域缺乏杂合子[21]。在基因组某一段区域内,当一定数量一定密度的SNPs表现为纯合时,可以判断该区域存在ROH现象。遗传漂变(Genetic Drift)、群体结构(Population Structure)、人工选择(Artificial Selection)、连锁不平衡(linkage Disequihbnum,LD)、近亲交配( inbreeding)等都会影响ROH的产生,其中近亲交配是影响ROH的首要因素[21,22]。不同的动物群体由于有着不同的群体大小、群体结构和交配体制,受到不同的自然或人工选择方式、选择强度影响,通过长期的历史进化过程,都会在基因组上形成独特的ROH模式,ROH可以反映不同动物群体所蕴含的独特遗传背景信息。ROH可以用于估计动物的近交情况[23,24]、推测近交的历史[25,26]、鉴定受到选择的基因[27,28]和有害突变[29-31]、评估遗传多样性和遗传资源保护[23,32-34],以及优化动物育种规划[29,35]。长的ROH片段反映最近世代发生过近交,而短的ROH说明较远世代产生近交,因为世代数越短,ROH片段被重组打断的可能性就越小。利用ROH计算基因组近交系数FROH可用于对物种或群体的近交情况进行评估,其计算方法为基因组中ROH片段的总长度占基因组总长度的比例。基因组近交系数FROH和系谱近交系数FPED呈中等程度相关或强相关,且基于ROH计算的基因组近交系数FROH最接近真实的近交系数[35-39]。
连续纯合子区域分析结果显示:新疆细毛羊、策勒黑羊、巴音布鲁克羊、阿勒泰羊的平均ROH长度分别约为110.665、93.532、88.417 和74.445 Mb,基因组近交系数分别约为0.042 3、0.035 8、0.033 8、0.028 5。新疆细毛羊在<0.5 Mb范围的ROH总长度显著高于巴音布鲁克羊、策勒黑羊和阿勒泰羊(P<0.05)。新疆细毛羊的平均ROH片段数量(215.8)显著高于策勒黑羊(166.2)、巴音布鲁克羊(152.9)和阿勒泰羊(150.2)(P<0.05),4个绵羊群体遗传多样性顺序为:阿勒泰羊>巴音布鲁克羊>策勒黑羊>新疆细毛羊。
3.2.3 连锁不平衡分析
连锁不平衡(Linkage Disequilibrium,LD)是指分属两个或两个以上基因座位的等位基因同时出现在一条染色体上的几率,高于随机出现的频率,呈现出一种相互关联的现象[40,41]。重组率(Recombination),遗传漂变(Genetic Drift),突变速率(Mutation Rate),自然选择(Natural Selection),交配行为方式(Mating Behavior),遗传连锁方式(Genetic Linkage Mode)等许多因素都会对连锁不平衡产生影响。不同物种或群体所经历的特殊历史事件及繁殖行为等特征可以通过连锁不平衡情况进行反映。驯化选择,会导致群体遗传多样性下降,位点间的相关性(连锁程度)加强。通常驯化程度越高,选择强度越大的群体,LD衰减速度是最慢的。连锁不平衡分析结果表明:阿勒泰羊的连锁不平衡程度相对最低,新疆细毛羊的连锁程度相对最高的,其次是策勒黑羊和巴音布鲁克羊,该结果与杂合度和连续纯合子区域分析的结果基本一致,阿勒泰的遗传多样性最高,其次是巴音布鲁克羊,新疆细毛羊的遗传多样性最低。
4 结 论
新疆细毛羊的平均观测杂合度(Ho=0.196)极显著低于阿勒泰羊(Ho=0.222)(P<0.001),低于巴音布鲁克羊(Ho=0.201),略高于策勒黑羊(Ho=0.194);新疆细毛羊在<0.5Mb范围的ROH总长度显著高于巴音布鲁克羊、策勒黑羊和阿勒泰羊(P<0.05),其平均ROH片段数量(215.8)显著高于策勒黑羊(166.2)、巴音布鲁克羊(152.9)和阿勒泰羊(150.2)(P<0.05);在4个绵羊群体中,新疆细毛羊的连锁不平衡程度相对最低。新疆细毛羊的遗传多样性水平相对低于阿勒泰羊、巴音布鲁克羊和策勒黑羊。
猜你喜欢
现代畜牧科技(2021年10期)2021-11-19
中国饲料(2021年17期)2021-11-02
花火·绘阅读(2021年7期)2021-08-21
种子(2021年3期)2021-04-12
四川文学(2020年11期)2020-02-06
小学阅读指南·低年级版(2017年4期)2017-04-24
外语教学理论与实践(2016年1期)2016-06-11
中国畜牧兽医文摘(2015年9期)2015-12-29
华东理工大学学报(自然科学版)(2014年1期)2014-02-27
山西农业大学学报(自然科学版)(2011年3期)2011-04-25
