
西藏黑木耳高产菌种不同生长发育时期转录组测序分析
2023-07-21 05:33张彦龙曾伟民
黑龙江大学自然科学学报 2023年3期
薛 娇, 于 洋, 张彦龙, 雷 虹, 曾伟民
(1.黑龙江大学 农业微生物技术教育部工程研究中心, 哈尔滨 150500; 2.黑龙江大学 生命科学学院, 哈尔滨 150080)
0 引 言
黑木耳属于真菌界,担子菌亚门,层菌纲,木耳目,木耳科,木耳属[1]。黑木耳口感脆嫩,味道鲜美可口,我国对其的食用已有上千年的历史。黑木耳子实体营养丰富,其多糖具有降血脂、抗血栓和抗肿瘤等作用[2],黑色素能够明显改善四氯化碳造成的急性肝损伤,多酚类和黄酮类物质具有抗氧化和清除自由基等功能。黑木耳在我国种植广泛,在云南、贵州、新疆、华北、内蒙古、西藏及东北地区均有分布。西藏黑木耳由于生长位置海拔较高和紫外线辐射较强的独特地理条件而具有抗逆性强、耐低温、生长周期短和营养价值高的优点[3]。
转录组在广义上是指整个细胞在某一特定条件或状态下所转录出的编码RNA和非编码RNA的总和,转录组学是在整体水平上研究生物个体所转录出RNA的差异情况[4]。转录组测序是通过将RNA进行测序,获得表达差异基因,注释到各个数据库从而进行分析。目前,用于转录组研究的主要有SAGE基因表达序列分析技术、大规模平行测序技术、转录组测序技术和基因芯片技术[5]。转录组测序技术并不局限于对已知转录本信息的样品进行检测,还可以利用高通量测序对无参考序列信息的样品进行检测。近年来随着测序成本的降低和其灵敏度高、检测范围广和重复性好的特点被广泛应用到各个领域。王思雨等对成菇期和变色期鸡腿菇子实体进行转录组测序分析,发现变色期DNA复制、转录和翻译能力下降,这可能与鸡腿菇自溶现象有关[6]。刘璐等通过对羊肚菌正常菌丝和退化菌丝进行转录组测序,筛选出一些差异表达基因,注释后得出羊肚菌菌丝退化与代谢过程有关,为后续解决菌丝退化问题提供参考[7]。赵震宇等通过对不同浓度单宁处理过的玉木耳菌丝进行转录组测序,得到2 012个差异基因,为进一步研究单宁对玉木耳菌丝的影响机制提供参考[8]。本试验采用课题组分离选育出的西藏黑木耳高产菌种,对其子实体3个不同发育阶段转录组进行分析研究。对黑木耳子实体的unigene使用生物信息学分析工具,进行功能注释和分析。该研究为下一步筛选和挖掘有关黑木耳子实体生长发育的功能基因提供重要数据参考,以及为黑木耳品种改良提供了科学依据。
1 材料与方法
1.1 材料与仪器
1.1.1供试菌株
西藏黑木耳高产菌种(专利号:ZL 2013 1 0278146.8)由本实验室前期对比选育分离得到,命名为西藏6号[3]。
1.1.2样品的采集
黑木耳子实体采摘自黑龙江省勃利县黑木耳种植基地,在黑木耳子实体生长的三个发育阶段(幼耳期、旺盛期和成熟期),采取无杂菌、无污染、品质优良的黑木耳置于放有冰袋的泡沫盒中,立即运至实验室,转移至冻存管后,使用液氮将样品速冻0.5 h,置于-80 ℃超低温冰箱中用于RNA提取,委托上海美吉生物医药科技有限公司测序。
1.1.3仪器
DU530紫外可见分光光度计(美国Beckman公司);DYY-12电泳仪(北京六一生物科技有限公司);DYCZ-24DN水平电泳槽(北京六一生物科技有限公司);Agilent 2100生物芯片分析系统(美国Agilent科技有限公司);Illumina NovaSeq 6000测序系统(美国Illumina公司);Elite超低温冰箱(美国Revco公司)。
1.2 试验方法
1.2.1总RNA样品提取
分别在西藏6号黑木耳幼耳期、旺盛期和成熟期取样,每个样品设置3个平行实验,共9个样品。每个样品取50 mg,分别加入250 μL NucleoZOL裂解细胞组织,再加入200 μL RNase-free的水沉淀污染物,分别用250 μL异丙醇和250 μL 75%乙醇沉淀和洗涤RNA。用NanoDrop[9]检测RNA吸收峰是否正常,利用琼脂糖凝胶电泳对提取到的待测样品RNA进行检测,使用Agilent 2100[10]精确检测RNA的RIN值、28S/18S和5S峰。所有指标均需符合测序的质量要求。
1.2.2RNA测序
提取后的RNA为总RNA,需要对其中的mRNA进行富集。将碱基末端带有T的磁珠与测序RNA 3′末端的ployA结合,加入buffer将富集后的mRNA打成短片段。以mRNA为模板,加入六碱基随机引物和逆转录酶,合成单链cDNA,再加入dDNTPs、DNA聚合酶和缓冲液合成第二条链,最后加A尾,PCR扩增15个循环,对文库进行测序[11]。RNA测序服务由上海美吉生物医药科技有限公司提供。
1.2.3转录组测序数据处理
测量后的数据中包含测序接头序列、不确定碱基信息N率较高的序列、一些低质量的读段片段和一些影响分析的长度过短的片段,这些因素都会影响后续分析结果的准确性。对于需要去除接头序列,并且序列中含有质量小于10的碱基的reads,整条序列都需要剔除。为了得到高质量的有效读数片段,需要剔除不确定碱基信息N率大于10%的片段和修剪后片段长度小于30 bp的序列。本研究无参考基因组,利用Trinity[12](https://github.com/trinityrnaseq/trinityrnaseq)将所有质量控制后的测序数据进行组装,组装后利用TransRate[13](http://hibberdlab.com/transrate/)和CD-HIT[14](http://weizhongli-lab.org/cd-hit/)对组装结果进行优化过滤,利用BUSCO[15](Benchmarking universal single-copy orthologs, http://busco.ezlab.org)对组装结果进行评估,评估基因组或转录组的组装完整性。取测序数据最长的转录本作为unigene进行后续分析。
1.2.4差异表达基因筛选
使用DESeq2软件[16]对Unigene进行分析,以筛选差异表达基因,FDR<0.05的基因为显著差异基因。
1.2.5Unigene功能注释
将所有的Unigene与六大数据库(GO、KEGG、COG、Nr、Swiss-Prot和Pfam)进行比对,获得Unigene和Transcript的注释情况,并对各注释信息进行统计分析。
1.2.6GO和KEGG功能富集分析
利用Goatools软件[17](https://github.com/tanghaibao/GOatools)和R语言编写脚本对基因集中的转录本进行GO和KEGG富集分析。GO和KEGG通路富集均默认,P值(Pvalue_corrected)<0.05时,GO和KEGG通路存在显著富集情况。
2 结果与分析
2.1 测序数据质量控制
黑木耳子实体转录组测序数据如表1所示。由表1可以看出,从西藏黑木耳的9个样品中共获得51 482 040~77 672 734条统计过滤后测序数据,Clean bases为7 685 784 278~11 565 940 347 G。样品测序的Q20含量超过98%,GC含量为60.54%~61.48%,Q30碱基含量均不小于94.84%。根据结果对比统计得出,各样品的Reads测序错误率为0.023 9~0.024 3。熊宇晴等对无患子叶片进行高通量测序后,得出数据质量情况,GC占总碱基数的42.73%,Q20和Q30含量分别为97.79%和94.00%,符合下一步的组装要求[18]。从西藏6号黑木耳质控数据结果得出,样品测序结果较好,符合转录组进行组装和注释的要求。

表1 质控数据表
2.2 转录组从头组装结果
对Transcript有效读取片段进行组装拼接,去掉其中冗杂多余的片段,共生成28 817个Transcript(表2),组装得到的所有Transcript碱基为36 006 442个,平均长度为1 249.49 bp。Unigene共有13 765个,将所有的Unigene组装得到的碱基为15 530 526个,平均长度为1 128.3 bp。西藏6号黑木耳Unigene的N50数值为1 954。在物种差异、拼接算法和测序平台都正确的前提下,N50数值能够作为评估序列测度的重要参数,反映拼接所得序列的完整程度。E90N50长度分别为2 891和2 640。所有样本质控后的有效数据合并后与组装Unigene/Transcript进行比较,获得的mapped率分别为59.606%和66.171%。总碱基数量中的58.19%和58.91%是GC碱基。李方东利用BUSCO对茶树中直系同源单拷贝基因完整性进行考察,以评价转录本组装的准确性和完整性,相似性评分越高,组装结果越好[19]。用BUSCO评估Unigene和Transcript,得分分别为83.5%和90.0%,表明组装结果完整性和准确性较高。
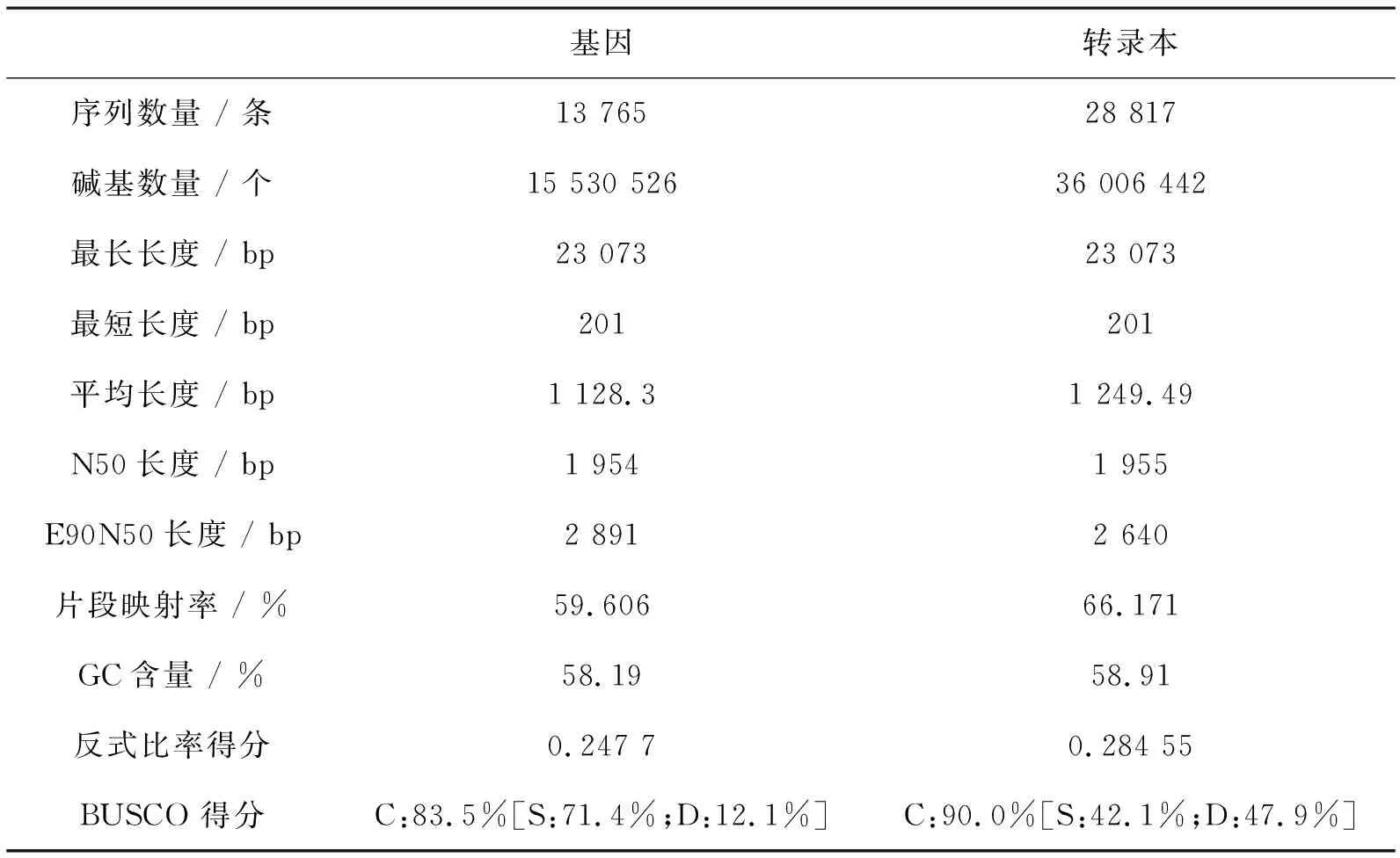
表2 优化组装结果评估表
如表3所示,过滤后测序数据为25 741 020~38 836 367条,可以对比到组装Transcript上的过滤后测序数据为17 330 951~27 367 399条,可以追踪到组装Transcript上的过滤后测序数据所占百分比为67.03%~70.47%。

表3 测序数据与组装结果对比
2.3 差异表达基因分析
表4为差异基因数目统计表,从获得的差异表达基因集的基因数目统计得出,西藏黑木耳子实体旺盛期和成熟期差异基因为1 225个,上调基因为661个,下调基因为564个。幼耳期和旺盛期差异基因为2 096个,比旺盛期和成熟期的差异基因多,同时上调基因也比下调基因表达得更多,上调基因为1 225个,下调基因为871个。

表4 差异基因数目统计表

图1 差异基因数目统计图(a)和维恩图(b)
图1为差异基因数目的统计图和信息图。维恩图显示,西藏6号黑木耳幼耳期、旺盛期和成熟期3个不同发育时期的共同差异基因有13 281个。其中,成熟期和旺盛期的差异基因有14 335个,幼耳期和成熟期的差异基因有13 717个,幼耳期和旺盛期的差异基因有14 093个。
2.4 功能注释及统计
将组装后的黑木耳子实体转录组序列与六大数据库进行对比,所有的Unigene和Transcript数量分别为27 529个和57 634个,表达的Unigene和Transcript数量分别为27 444个和57 406个,注释到数据库的表达Unigene和Transcript数量分别为18 258个和40 469个。在六大数据库上表达的Unigene分别注释了1 1967个(GO)、7 658个(KEGG)、5 252个(COG)、17 132个(Nr)、9 240个(Swiss-Prot)和11 328个(Pfam),表达的Transcript分别注释到27 232个(GO)、15 425个(KEGG)、9 090个(COG)、38 968个(NR)、19 555个(Swiss-Prot)和24 214个(Pfam)。

表5 转录本功能注释统计表
2.5 GO功能富集分析
对西藏6号黑木耳子实体幼耳期和旺盛期差异表达基因进行GO功能富集分析,结果如图2所示。基因集中的基因可以分成BP(Biological Process, 生物过程)、MF(Molecular Function, 分子功能)及CC(Cellular Component, 细胞组分)三个类别。在“生物过程”类别中,参与类异戊二烯生物合成过程(GO:0008299)的基因有6个,类异戊二烯代谢过程(GO:0006720)的基因有6个,谷氨酸脱羧成琥珀酸过程(GO:0006540)基因有3个,GO term显著富集,表明黑木耳子实体在生长发育和代谢调节等方面活动频繁。在“细胞组分”类别中,差异表达基因主要富集在膜的组成部分(GO:0016021)和膜的固有组分(GO:0031224),分别有415个。在“分子功能”类别中,最为富集的Unigene具有催化活性和结合活性。表明黑木耳子实体中大量的Unigene可以翻译为具有催化活性的酶和具有结合功能的蛋白。
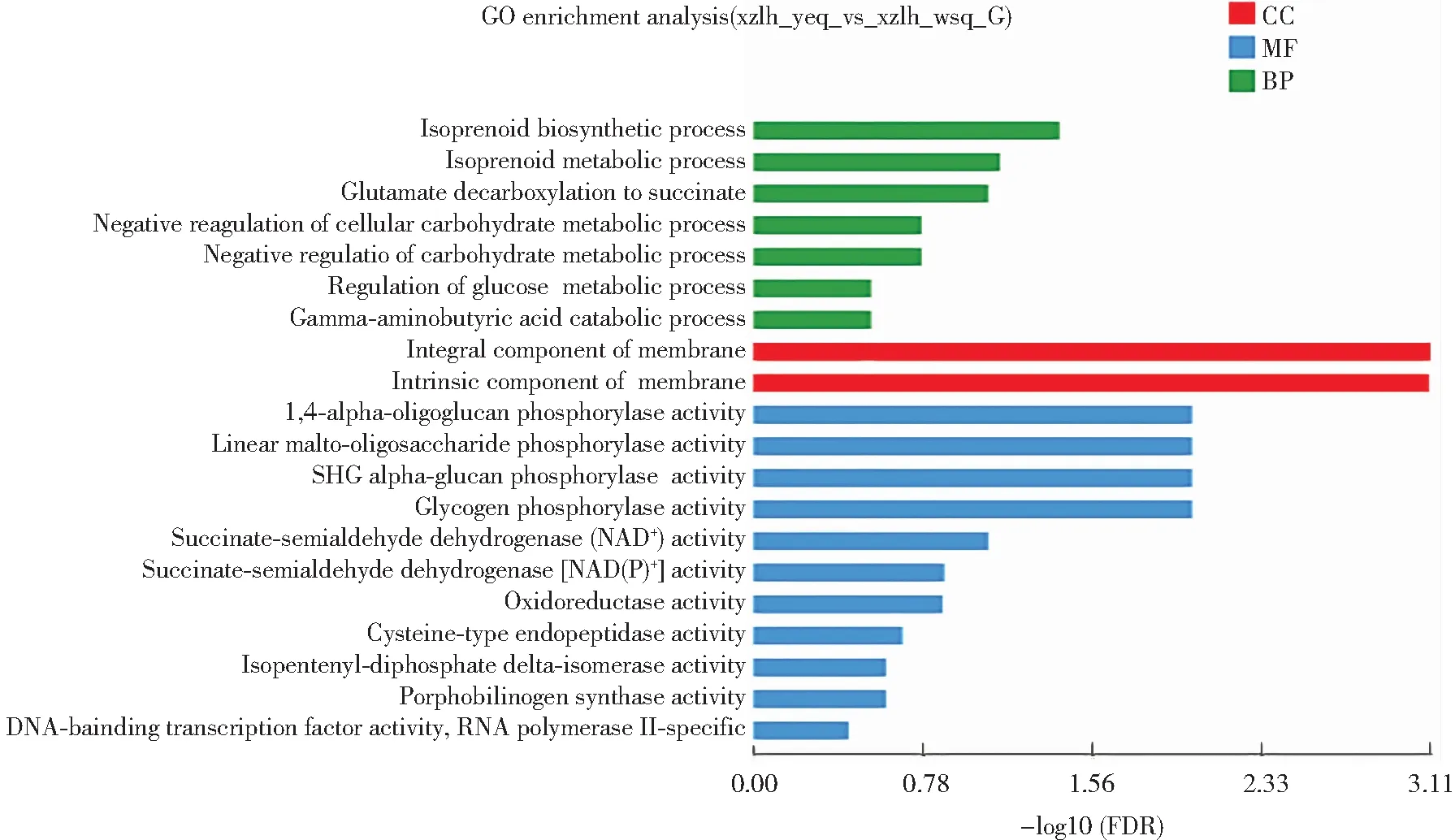
图2 黑木耳子实体幼耳期和旺盛期差异表达基因GO功能富集分析图
将黑木耳子实体旺盛期和成熟期的差异表达基因进行GO功能富集分析,结果如图3所示。在“细胞组分”类别中,参与膜的固有组分(GO:0031224)和膜的组成部分(GO:0016021)的差异表达基因分别有168个。在“分子功能”类别中,参与金属末端肽酶活性(GO:0004222)的基因有12个,参与肽链内切酶活性(GO:0004175)的基因有23个,表明黑木耳子实体中具有催化肽类活性的Unigene最为富集。从GO功能富集分析结果可以看出,在生物学过程类别中,差异表达基因主要富集在异类戊二烯的合成和代谢过程,说明差异表达基因在“生物学过程”主要参与细胞内进行的各种异类戊二烯的合成和代谢活动。在“细胞组分”类别中,富集到膜的固有组分和膜的组成部分的差异表达基因最多,表明细胞膜成分随着西藏6号黑木耳不断生长而发生改变。在“分子功能”类别中,在催化活性通路的差异表达基因最多。曾倩倩等对小麦叶片差异表达基因进行GO富集分析,检测到的1 207个DEGs富集到3个类别的34个条目下,主要参与催化、代谢和水解酶活性等,表明小麦受干旱环境的胁迫作用是多方面的[19]。
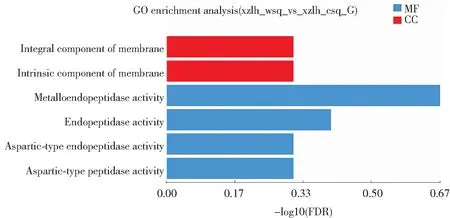
图3 黑木耳子实体旺盛期和成熟期差异表达基因GO功能富集分析图
2.6 KEGG功能富集分析
将西藏6号子实体幼耳期和旺盛期差异表达基因进行KEGG功能富集分析,结果如图4所示。富集到的Unigene有6个类别,20个亚类。在子类别中,p<0.05的有8个亚类。Unigene最为富集的途径是碳水化合物代谢(16%)、氨基酸代谢(15%)、脂质代谢(12%)、辅助因子和维生素代谢(9%)、其他氨基酸代谢(7%)以及运输和分解代谢(7%),说明西藏6号黑木耳子实体生长发育过程中碳代谢和氮代谢较为旺盛,为黑木耳子实体生长发育提供营养与能量。
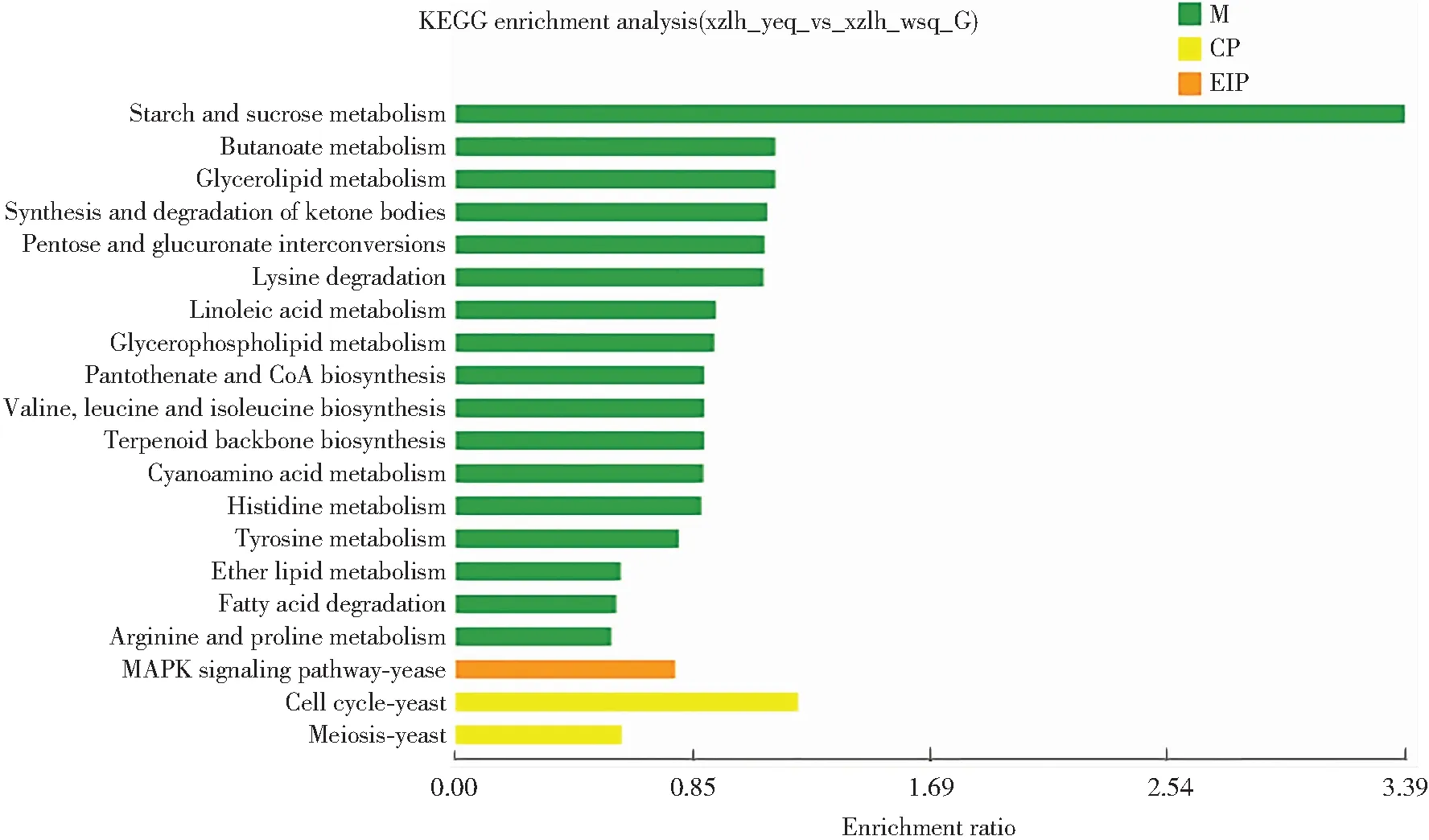
图4 黑木耳子实体幼耳期和旺盛期差异表达基因KEGG功能富集分析图
曾倩倩等对小麦叶片DEGs的KEGG通路富集发现,存在淀粉和蔗糖代谢、叶绿素代谢、光合作用、苯丙烷生物合成、甘油磷脂代谢、氨基酸代谢及光合生物中的碳固定等通路。表明植物受干旱环境的胁迫,光合作用-天线蛋白、叶绿体组分、氨基酸代谢和碳代谢等通路受到影响,光合作用和物质积累受到抑制,这可能与小麦叶片相对含水量降低及MDA含量增加有关[20]。卢园萍等通过对巴氏蘑菇子实体3个阶段的差异表达基因进行KEGG富集分析发现,原基期上调DEGs主要富集在核糖体蛋白和DNA复制,表明原基期细胞代谢旺盛,其中核糖体蛋白基因上调为后期蛋白质合成提供重要场所;采收期和开伞期差异表达基因主要富集在碳水化合物代谢、脂肪酸降解和氨基酸代谢等途径,为巴氏蘑菇子实体的生长发育与成熟提供营养和能量[21]。吴晓梅等对双孢蘑菇子实体3个发育时期进行KEGG富集分析,发现DEGs参与了氨基酸代谢、碳水化合物代谢、核苷酸代谢、脂类代谢和能量代谢这五大代谢通路,其中差异基因主要富集在氨基酸代谢通路中,氨基酸合成相关的多数基因上调表达,表明双孢蘑菇子实体发育形成需要一系列代谢反应协同调控,氨基酸代谢相关基因可能在双孢蘑菇子实体发育过程中起重要作用[22]。
将西藏6号黑木耳子实体旺盛期和成熟期差异表达基因进行KEGG功能富集分析,结果如图5所示。富集到的Unigene主要有6个类别,19个亚类。有5个亚类在子类别中p<0.05,Unigene最为富集的途径是氨基酸代谢(19%)、碳水化合物代谢(13%)、脂质代谢(12%)、聚糖的生物合成和代谢(9%)、运输和分解代谢(7%)、其他氨基酸代谢(6%)及能量代谢(4%)。表明黑木耳生长发育过程中代谢过程极其丰富,氨基酸代谢在黑木耳子实体生长中提供能量和营养。与吴晓梅等对双孢蘑菇子实体不同发育阶段转录组分析结果[21]类似,氨基酸代谢和碳水化合物代谢也有大量的差异基因富集。
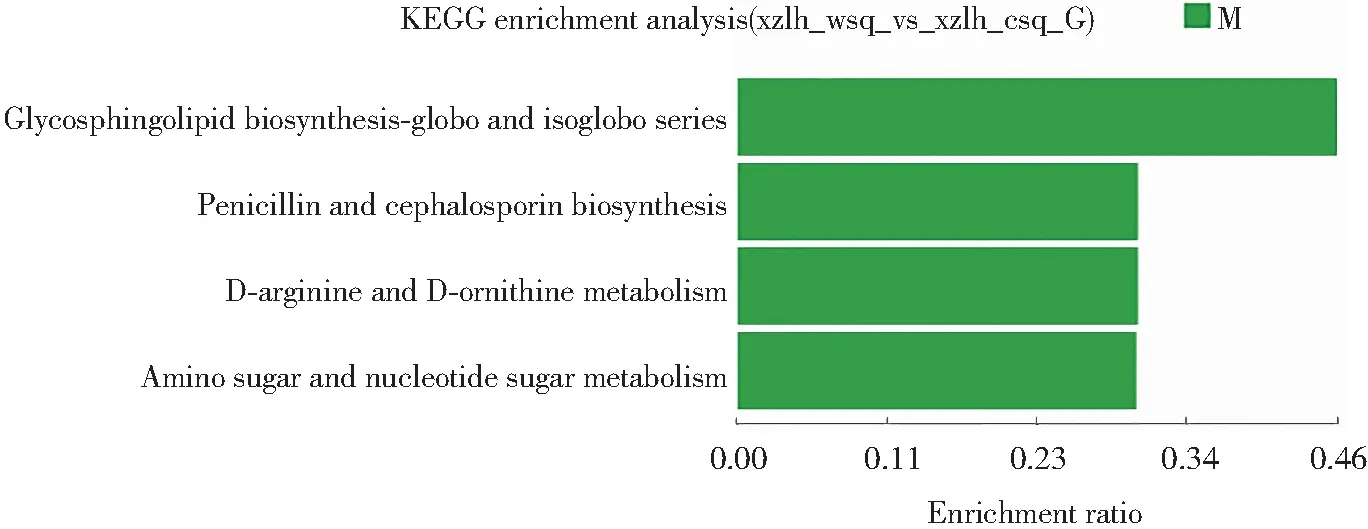
图5 黑木耳子实体旺盛期和成熟期差异表达基因KEGG功能富集分析图
3 结 论
对黑木耳子实体发育过程中3个不同阶段样品的转录组进行测序,并对转录组进行注释以及GO和KEGG功能富集分析。对黑木耳子实体9个样品的测序共获得 81.93 Gb Clean data,组装后得到Unigene为13 765个,Transcript为28 817个,平均长度为1 128.30 bp,N50长度为1 954 bp。以上结果表明,黑木耳子实体转录组测序和组装质量都较高。采用生物信息学进行分析,将组装后黑木耳子实体转录组序列与六大数据库进行对比,注释到数据库的Unigene共有18 258个,占全部Unigene的66.53%。注释到数据库的Transcript共有40 469个,占全部Transcript的70.5%。
从GO功能富集分析的结果可以看出,在“生物学过程”类别中,差异表达基因主要富集在异类戊二烯的合成和代谢过程,说明差异表达基因在生物学过程主要参与细胞内进行的各种异类戊二烯的合成和代谢活动。在“细胞组分”类别中,富集到膜的固有组分和膜的组成部分的差异表达基因最多,表明随着西藏6号黑木耳不断生长,细胞膜成分发生改变,细胞膜能够调节物质运输,维持细胞内环境的稳定。在“分子功能”类别中,在催化活性通路的差异表达基因最多。KEGG富集分析有利于了解基因的生物学功能,本研究对西藏6号黑木耳子实体3个不同时期发育阶段差异基因进行KEGG分析,筛选出多条与黑木耳子实体不同生长发育阶段有关的代谢途径,包括氨基酸代谢、碳水化合物代谢、脂质代谢和聚糖的生物合成和代谢等。表明黑木耳生长发育过程中代谢过程极其丰富,氨基酸代谢在黑木耳子实体生长中提供营养和能量。本研究结果为下一步筛选和挖掘有关西藏黑木耳子实体生长发育的功能基因提供重要数据参考,为黑木耳品种改良提供了科学依据。
猜你喜欢
学与玩(2022年7期)2022-10-31
心电与循环(2020年1期)2020-02-27
贵茶(2019年3期)2019-12-02
小猕猴智力画刊(2017年12期)2017-12-27
海峡姐妹(2017年4期)2017-05-04
剑南文学(2016年11期)2016-08-22
食药用菌(2016年6期)2016-03-01
西藏科技(2015年1期)2015-09-26
湖北农业科学(2014年3期)2014-07-21
云南中医学院学报(2011年4期)2011-07-31
