
儿童肝豆状核变性的临床资料与ATP7B基因分析
2023-07-31 03:26彭晓康刘小乖李瑞娜
中国妇幼健康研究 2023年7期
刘 攀,舒 畅,唐 丽,彭晓康,刘小乖,李瑞娜
(西安交通大学附属儿童医院感染科,陕西 西安 710003)
肝豆状核变性(Wilson′s disease,WD)是一种由ATP7B基因缺陷导致的铜代谢障碍性疾病[1-2],是目前少数可治疗的遗传代谢性疾病之一[3]。WD患病率为(0.25~4.00)/10 000,但理论发病率远高于此[4-5]。ATP7B基因编码的ATP7B蛋白属于高度保守的P型三磷酸腺苷(adenosine-triphosphate,ATP)酶超家族的1B类,负责将铜转运入高尔基体或毛细胆管内,在铜的排泄中起主要作用。ATP7B基因变异导致ATP7B酶活性改变,从而引起肝细胞内铜含量增加,造成肝细胞死亡和铜渗漏入血,进一步引起肝、脑、眼睛、肾脏等组织器官的损伤[2,4]。WD的临床表现复杂多样,可为无症状性转氨酶升高、急慢性肝炎、肝硬化、肝衰竭,也可表现为急性溶血性贫血、神经精神症状等[1,3-4]。本研究对近年来在西安交通大学附属儿童医院就诊的158例WD患儿的临床特征与ATP7B基因进行总结分析,以提高临床医师对该病的认识,并进一步探讨本地区WD患儿的遗传学特征。
1资料与方法
1.1研究对象
回顾性收集2016年1月至2022年6月期间收治于西安交通大学附属儿童医院感染科的158例诊断明确且为初诊的WD患儿作为研究对象。诊断标准:根据中华医学会肝病学会分会遗传代谢性肝病协作组制定的《肝豆状核变性诊疗指南(2022年版)》[4],推荐应用2001年莱比锡第8届WD国际会议的诊断标准即Leipzig评分系统[6],总分≥4分可确诊。本研究经西安市儿童医院伦理委员会批准(批准文号:论20220073),并豁免患儿及家属知情同意。
根据WD患儿的受累器官及临床表现的不同,分为肝型WD组和脑型WD组,分别有144例、14例。根据肝脏受累的轻重程度及病程长短、表现的差异,肝型WD组又分为症状前WD、急慢性肝炎型WD,分别有114例和30例。
1.2资料收集
通过我院电子病历系统,回顾性收集研究对象初诊时的临床表现、辅助检查,包括血尿常规、肝功、铜蓝蛋白(ceruloplasmin,CER)、24h尿铜、血清铜、凝血功能、腹部超声及头颅影像学、基因检测和眼科角膜K-F环等结果。
1.3 ATP7B基因检测
经患儿监护人签署书面知情同意书后,采集患儿及父母静脉血3mL,提取基因组DNA,并构建与代谢性肝病相关目标基因(ABCB4、ATP7B、CCDC115、NBAS、TMEM199等共573个基因)的全基因组文库。利用液相捕获试剂盒捕获目标基因后进行高通量测序,通过生物信息学分析找出相关基因的变异信息。当疑似有外显子缺失、临床高度怀疑WD但未检测出变异基因或仅检测到1个变异时,行多重连接依赖探针扩增或全外显子测序进一步分析。根据获得的变异结果对患儿及其父母进行Sanger测序验证。采用生物信息分析软件PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)、SIFT(http://sift.jcvi.org)及MutationTaster (http://mutationtaster.org)对新变异基因进行致病性预测。依据美国医学遗传学及基因组学学会(Ame-rican College of Medical Genetics and Genomics,ACMG)的遗传变异分类标准与指南[7-8],将变异基因归类为致病的、可能致病的、意义不明确的、可能良性的或良性的变异。
1.4统计学方法
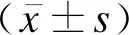
2结果
2.1一般情况
158例WD患儿中男性89名,女性69名,男女比为1.29∶1。WD患儿来自于148个非血缘关系家庭,17例有明确的肝病或WD家族史,其中6例为先证者的家系筛查中发现;确诊年龄为6.33(4.03~9.67)岁,其中最小诊断年龄为1.21岁,病程为1.00(0.42,4.75)个月。
症状前WD、急慢性肝炎型WD与脑型WD患儿的性别差异无统计学意义(P>0.05),症状前WD患儿的确诊年龄明显小于急慢性肝炎型WD与脑型WD(Z值分别为-56.640、-64.956,P<0.05),急慢性肝炎型WD患儿的病程短于症状前WD患儿(Z=26.930,P<0.05),见表1。
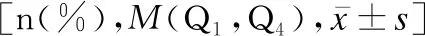
表1 肝型WD组与脑型WD组患儿的一般情况与临床表现比较
2.2临床表现
①以肝脏为首发表现的患儿(肝型WD组)有144例,占91.14%,其中无症状患儿114例,占72.15%,仅表现为铜代谢指标异常者有4例,转氨酶升高者110例;查体发现肝脏肿大者36例,其中4例伴脾脏肿大。98例患儿行眼科检查,其中5例存在角膜K-F环。②以急性慢性肝炎表现者30例,均有转氨酶升高,其中28例出现不同程度的黄疸(27例为胆汁淤积,1例入院时为高胆红素血症、住院第2天复查转为胆汁淤积),24例存在腹胀或腹痛,21例存在腹腔积液,21例出现肝脏肿大、肝硬化,20例伴有脾脏肿大,21例出现急性肝衰竭,其中18例伴有Coomb′s阴性溶血性贫血。29例患儿行眼科检查,其中24例存在角膜K-F环。见表1。
以神经精神症状为主要表现的WD(脑型WD)患儿有14例,10例有吐字不清、流涎,8例有肢体抖动、震颤,4例有肌张力增高,3例有步态异常,3例有吞咽、咀嚼障碍,3例有情绪低落、表情淡漠,2例有轻度黄疸,2例有转氨酶轻至中度升高。13例患儿行眼科检查,均存在角膜K-F环。
症状前WD、急慢性肝炎型WD与脑型WD患儿在是否存在K-F环、转氨酶升高、黄疸、腹胀或腹痛、肝脏肿大、脾脏肿大、腹腔积液、肝硬化、溶血性贫血及神经精神症状方面差异均有统计学意义(P<0.05),见表1。
2.3实验室检查
158例WD患儿的CER值为0.03(0.01,0.06)g/L,24h尿铜为177.00(115.00,289.60)μg/24h,血清铜为9.87(7.36,13.19)μmol/L。症状前WD、急慢性肝炎型WD、脑型WD患儿的总胆红素(total bilirubin,TB)、直接胆红素(direct bilirubin,DB)、丙氨酸氨基转移酶(alanine aminotransferase,ALT)、尿铜、血小板(platelet,PLT)差异均有统计学意义(H值分别为70.150、68.443、60.504、39.502、51.343,P<0.05),见表2。进一步两两比较发现:症状前WD患儿的血清铜水平较急慢性肝炎型WD患儿低(Z=-27.313,P<0.05),24h尿铜较急慢性肝炎型WD、脑型WD患儿低(Z值分别为-54.929、-33.005,P<0.05)。脑型WD患儿的天冬氨酸氨基转移酶(aspartate aminotransferase,AST)、白细胞(white blood cell,WBC)较症状前WD与急慢性肝炎型WD患儿低(AST:Z值分别为51.234、52.013;WBC:Z值分别为39.033、38.981,P<0.05)。急慢性肝炎型WD患儿的血清白蛋白(albumin,Alb)、碱性磷酸酶(alkaline phosphatase,ALP)、红细胞计数(red blood cells,RBC)、血红蛋白(hemoglobin,Hb)较症状前WD、脑型WD患儿低(Alb:Z值分别为69.710、-40.250;ALP:Z值分别为54.067、-34.673;RBC:Z值分别为64.734、-52.923;Hb:Z值分别为62.763、-64.000,P<0.05);CER水平、γ-谷氨酰转肽酶(γ-glutamyl transpeptidase,GGT)、较症状前WD、脑型WD患儿高(CER:Z值分别为-32.257、36.000;GGT:Z值分别为-32.571、54.154,P<0.05)。
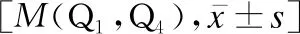
表2 肝型WD与脑型WD组患儿的实验室检查比较
2.4 ATP7B基因
158例WD患儿中有116例进行了基因检测,基因检测率为73.42%,症状前WD患儿90例,急慢性肝炎型WD患儿21例,脑型WD患儿5例;其中5例为杂合变异,14例为纯合变异,97例为复合杂合变异(8例有3种变异类型)。共检测出64种变异,包括错义突变39种、同义变异3种、无义变异4种、移码变异8种、剪切变异5种、缺失变异5种(1种单个氨基酸缺失,4种外显子缺失)。热点突变前4位依次为c.2333G>T(p.R778L)、c.2621C>T(p.A874V)、c.2310C>G(L770=)及c.2975C>T(p.P992L),等位基因频率分别为28.88%、9.48%、8.62%、7.76%。发现6种新的未报道的基因变异,包括c.1908dupC(p.Asn637Glnfs*118)、c.4179_4180insC(p.Pro1394 Profs*15)、c.1604A>G(p.Glu535Gly)、c.2278C>T(p.Pro760Ser)、c.3008C>A(p.Ala1003Glu)及c.3532A>C(p.Thr1178Pro)。根据ACMG指南判定,除c.1604A>G(p.Glu535Gly)为意义不明的变异外,其余均为可能致病性变异。
依据患儿是否存在c.2333G>T(p.R778L)变异进行基因型的分组,分为R778L型WD与其他型WD,分别有55例、61例。R778L型WD的确诊年龄大于其他型WD(Z=-2.082,P<0.05),而临床分型、病程、ALT、AST、WBC、Hb、PLT、CER、尿铜、血清铜、K-F环阳性率两组比较差异均无统计学意义(P>0.05),见表3。

表3 R778L型WD与其他型WD患儿的临床比较 [n(%),M(Q1,Q4)]
3讨论
3.1儿童WD的一般情况与临床表现
WD是一种铜蓄积性的遗传代谢病,常以肝脏受累起病,发病年龄相对较早[3]。我院收治的WD患儿中肝型WD占91.14%,其中以仅表现为肝酶升高,伴或不伴有肝肿大的症状前WD居多,占72.15%。症状前WD患儿多在入园、入学体检时发现,确诊年龄中位数为5.30岁,明显小于急慢性肝炎型WD与脑型WD的确诊年龄(P<0.05)。WD最小诊断年龄为1.21岁,为先证者的家系筛查中发现。WD患儿基因检测率为73.42%,有部分患儿监护人认为临床已明确诊断为WD而不需进行基因检测,这是错误的观点,仍推荐对先证者的直系亲属或有WD家族史者进行家系的基因检测[4,9-11]。
急慢性肝炎型WD组中有21例急性起病,出现肝功能衰竭,其中18例伴有Coomb′s阴性溶血性贫血。虽然患儿黄疸、腹痛或腹胀等病程短(中位数为0.37个月),但查体多有慢性肝病的异常体征,如肝硬化、腹水等。WD患儿,当肝细胞内的铜超载到一定水平会造成肝脏和其他组织器官损害,但肝脏为沉默器官,无明显临床症状时多不会引起患儿不适或家长关注,往往就诊时已出现代偿期甚至失代偿期肝硬化。这与Ferenci等人[12]报道的39.50%的儿童及青少年在诊断WD时已存在肝硬化的结果一致。2例患儿出现表情淡漠、震颤的神经精神症状,结合血氨异常升高,头颅磁共振检查正常,诊断为高血氨症、肝性脑病。对于肝型WD伴随有神经精神症状的患儿,临床医师需注意鉴别肝性脑病与WD神经系统损害表现,以免贻误病情。
3.2儿童WD的辅助检查
急慢性肝炎型WD的CER、GGT显著高于症状前WD及脑型WD组,而ALP、ALB、RBC、Hb均较另外两组明显下降。急慢性肝炎时肝细胞损伤、细胞膜破坏,细胞内蓄积过量的铜离子、铜蓝蛋白前体被释放入血,铜离子的金属毒性对红细胞破坏可造成贫血,肾脏排泄铜的代偿作用加强,尿铜升高明显;肝损害造成肝脏合成功能下降,从而导致白蛋白降低。以急性肝衰竭起病的WD患者,铜蓝蛋白可能下降不明显或正常,早期诊断存在一定困难,临床医师仍需提高对WD诊断的警惕性[3,13]。有学者提出,在急性肝衰竭人群中识别WD的顺序指标:ALP/TB<4,AST/ALT>2.2,敏感度为94.00%,特异度为86.00%~94.00%[14],但其诊断效能仍有争议。
3.3儿童WD临床表型与基因表型的相关性分析
WD患者的ATP7B基因在不同地域、种族和人群的变异特征存在明显差异,欧洲以p.H1069G为主[15],而亚洲人群以p.R778L最为常见,等位基因频率为12.00%~39.00%[16-19]。我国WD患者主要以p.R778L、p.P992L和p.T935M变异为主[18],但不同地区的中国人也有不同的ATP7B变异类型,具有区域特异性分布的特征[20]。本研究WD患儿最常见的突变位点为c.2333G>T(p.R778L),等位基因频率为28.88%;其次为c.2621C>T(p.A874V),等位基因频率为9.48%,该发现与其他研究报道的中国WD人群的常见突变基因结果相一致[21-22]。
WD患儿的临床表现多种多样,临床表型差异较大,国内外众多研究均尝试探索WD的临床表型与基因型的相关性,但目前尚无统一且公认的结论出现。本研究发现,p.R778L型WD的确诊年龄高于其他型WD,而在临床分型、病程、CER、血清铜等方面两组差异无统计学意义。该结论与黄帆等[23]报道的“p.R778L突变WD患者的发病年龄较晚,与首发症状和铜代谢障碍无关”相符,而与Cheng等[18]报道的“中国WD患者中p.R778L组发病年龄更早,CER水平及血清铜更低”的结论不一致。WD临床表型与基因型是否相关存在一定争议,可能与不同地域、样本量的选取及修饰基因的影响作用等因素有关,仍待大量临床数据积累,以及多中心、大样本研究的验证。
利益冲突声明:所有作者均声明不存在利益冲突。
猜你喜欢
青春期健康(2022年14期)2022-08-02
天津医科大学学报(2021年4期)2021-08-21
青春期健康(2021年14期)2021-08-03
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中国生殖健康(2019年7期)2019-01-06
中国生殖健康(2019年7期)2019-01-06
中国卫生(2015年12期)2015-11-10
百科知识(2015年18期)2015-09-10
郑州大学学报(医学版)(2015年2期)2015-02-27
