
基于SNP芯片数据分析不同奶牛场基因组近交系数及筛选功能性基因
2023-07-31 08:57王振宇张赛博刘文慧任小丽闫跃飞高腾云黄河天
畜牧兽医学报 2023年7期
王振宇,张赛博,刘文慧,梁 栋,任小丽,闫 磊,闫跃飞,高腾云,张 震,3*,黄河天*
(1.河南农业大学动物科技学院,郑州 450046;2.河南省奶牛生产性能测定中心,郑州 450045;3.河南省种业发展中心,郑州 450046)
基因组长纯合片段(runs of homozygosity, ROH)一般存在于二倍体生物中,它是亲代将单倍型基因中同源相同(identity by descent, IBD)的片段遗传给子代,并且在子代的基因组中形成连续性的纯合片段[1],即子代从亲代继承了同源的染色体片段,从而导致后代基因组中的纯合片段产生并上升到ROH[2]。连锁不平衡、种群瓶颈、遗传漂变、近亲交配和选择都可能是引起ROH产生的因素[1,3-4]。不同的群体历史会产生不同长短的ROH,长片段ROH通常由群体近几个世代近交产生,短片段ROH通常来自更远的祖先[5-7]。因此,通过全基因组ROH特征的检测,可以了解种群历史、结构、近交情况。
ROH最早在人类染色体基因组发现,并被认为可能对人类健康有重要影响。随着ROH在人类群体遗传学中研究的深入[8-10],不同畜禽的ROH分析研究也逐渐开展[11-13]。基于ROH估计基因组近交系数已成为利用全基因组信息评估近交的常用方法,即利用ROH计算基因组近交系数FROH(inbreeding calculated from ROH),它可以准确计算个体近交系数。现已有多项研究证明了基于系谱信息计算的近交系数要低于真实的近交系数。杨湛澄等[14]利用牛54K SNP芯片数据对北京地区2 107头荷斯坦牛基因组ROH分布进行了统计,并计算了基因组近交系数和系谱近交系数,发现基于ROH计算的基因组近交系数能更准确地反映个体的真实近交情况。Peripolli等[15]利用770K SNP芯片数据比较了2 908头吉尔牛(Gyr)基于ROH(FROH)、基因组关系矩阵(genomic relationship matrix,FGRM)、基因组纯合子百分比(homozygosity,FHOM)、系谱信息(pedigree,FPED)4种方法计算的近交系数,结果表明在没有系谱记录的情况下,FROH可用作近交估计的替代方法。此外,通过识别群体的高频ROH片段,鉴定到了与产奶量、乳成分、热适应相关的基因。Nani和Peagaricano[16]研究发现,基因组ROH与荷斯坦公牛繁殖性状显著相关,公牛群体中高度纯合的基因组区域与公牛繁殖性状呈现负相关,并在低繁殖力公牛ROH富集区域鉴定到与精子生物学和雄性生育能力密切相关的基因。Liu等[17]利用简化基因组测序的方法,通过ROH与综合单倍型评分(integrated haplotype score, iHS)分析,检测到与上海荷斯坦奶牛群体健康、繁殖、环境适应等有关的候选基因。通过对全基因组ROH进行检测,可以更准确地掌握群体的近交程度,帮助研究者在育种实践中制定科学合理的选种选配方案。鉴定全基因组的ROH也可以更好的了解ROH在染色体上的分布规律,进而挖掘可能影响畜禽重要性状的候选基因[18-20]。
在我国,北京[14]、上海[17]、宁夏[21]基于荷斯坦牛群体基因组ROH估算群体近交系数、检测与经济性状相关候选基因及选育过程中的选择信号等的研究,为中国荷斯坦奶牛育种提供了重要数据参考。然而,通过基因组ROH信息估计不同牧场荷斯坦奶牛群体近交水平和检测群体选择特征的研究仍然较少。本研究旨在利用奶牛150K SNP芯片数据对河南省7个奶牛场荷斯坦牛进行全基因组ROH检测,计算ROH的长度、频率、数目和分布以及基因组近交系数FROH,比较不同牧场荷斯坦牛基因组近交程度,并在高频ROH区域注释与荷斯坦牛经济性状相关的候选基因。以期为详细了解河南省荷斯坦牛群体基因组ROH分布特征及基因组近交程度,为牧场今后选种选配提供参考。也可通过ROH富集区域鉴定一些与奶牛经济性状相关的基因,为奶牛标记辅助选择提供候选基因信息,为奶牛场科学选种选配提供指导。
1 材料与方法
1.1 试验动物
根据系谱、生产数据记录的完整性,筛选出7个存栏量在150~5 000头的规模化牧场,按存栏量10%的比例抽取牧场核心群个体进行血液样本采集,最终共采集了900头荷斯坦牛。具体样本分布情况详见表1。

表1 不同奶牛场荷斯坦牛ROH长度和数量Table 1 The mean length and number of runs of homozygosity(ROH) in Holstein of different dairy farms
1.2 SNP芯片分型及数据质量控制
采集尾椎静脉血,提取DNA,利用GGP Bovine 150K芯片进行基因分型。用PLINK(v1.90)[22]对原始数据进行质控,设定条件:1)SNP检出率大于95%;2)个体检出率大于99%;3)最小等位基因频率大于0.01;4)哈迪-温伯格平衡P值大于10-6;5)保留常染色体数据。
1.3 群体结构及连锁不平衡分析
基于SNP信息,使用GCTA(v1.93)软件[23]对900头荷斯坦牛群体进行主成分分析(principal component analysis, PCA)。采用PopLDdecay(v3.42)软件[24]计算每个牧场的连锁不平衡(linkage disequilibrium, LD)程度,并使用软件自带的Plot_MultiPop.pl脚本绘制LD衰减曲线图。
1.4 ROH检测及基因组近交系数的计算
ROH检测使用PLINK软件[22],使用滑动窗口的方法对常染色体进行检测,具体检测参数如下:1)滑动窗口阈值使用0.05;2)滑动窗口设置50个SNPs位点;3)每一个滑动窗口中允许丢失的基因型为5个;4)每一个滑动窗口中允许的杂合子数目为1个;5)组成ROH的SNP的最大间隔为1 Mb;6)组成ROH的SNP的最低密度为每50 kb 1个SNP;7)ROH片段的最小长度设为500 kb;8)每个ROH至少由50个SNPs组成。
利用ROH计算近交系数(FROH),公式如下:
其中,∑LROH为常染色体上ROH片段长度之和,Lgenome为常染色体基因组物理长度之和(2.49 Gb)。
1.5 高频ROH区域候选基因鉴定
使用R语言统计每个SNP在奶牛群体中参与组成ROH的次数占样本数的比例,并将前1%的SNPs区域作为高频的ROH区域。基于高频ROH区段的物理位置,并通过生物数据库Ensembl[25]中的BioMart模块与牛参考基因组(Bos_taurus.ARS-UCD1.2)进行比对,检索基因,然后依据NCBI(https://www.ncbi.nlm.nih.gov/)、GeneCards(https://www.genecards.org/)网站及文献查询基因功能。运用KOBAS(http://bioinfo.org/kobas/)[26]在线数据库对注释到的基因进行KEGG通路富集分析,当P<0.05时,则表示显著富集。
2 结 果
2.1 SNP质控结果及群体遗传结构和连锁不平衡分析
在质控后每个个体保留了96 789个SNPs位点,相邻SNPs之间的平均距离为25.72 kb,以供后续分析。图1A显示了7个牧场荷斯坦牛群体的PCA分析结果。从图1可以看出,7个牛场主要分为了5个亚群。采用PopLDdecay分别计算各牧场群体的成对r2值,用于比较不同荷斯坦牛群体的LD水平(图1B)。LD分析显示,7个牧场奶牛群体LD衰减的顺序为:H7>H4&H5>H2&H3&H6>H1。
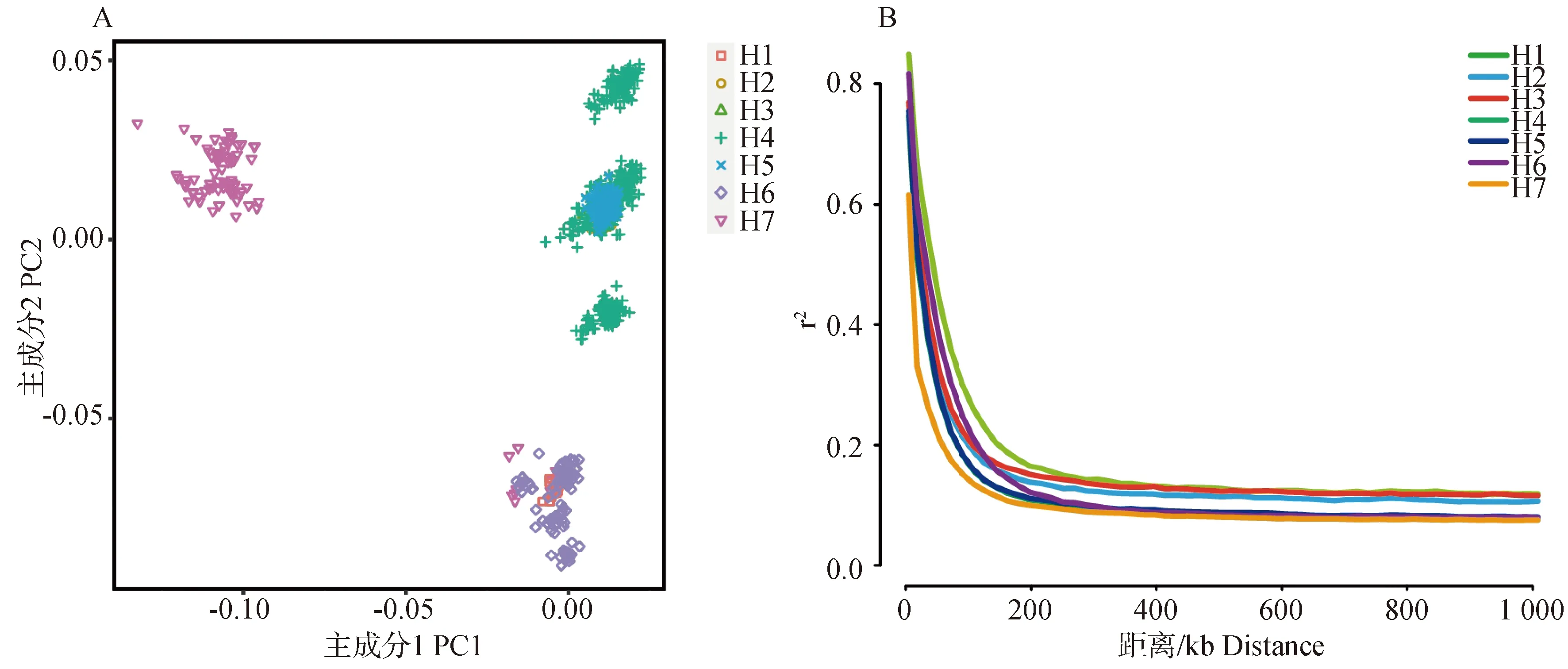
A.主成分分析图;B.LD衰减图。H1~H7代表牧场编号A.Principal component analysis of Holstein cattle population;B. LD decay of Holstein cattle population. H1-H7 represents pasture number图1 群体遗传结构及连锁不平衡Fig.1 Population genetic structure and linkage disequilibrium
2.2 ROH数目、长度及分布的统计
由表1可以看出,在7个牧场荷斯坦牛群体中共检测出55 908个ROH,ROH的平均长度为4.23 Mb,范围在1.90~14.07 Mb。其中H6号牛场ROH平均长度最小,为3.27 Mb;H2号牛场ROH平均长度最大为4.49 Mb。在0~5 Mb长度上,ROH总体比例占76.21%,其中H1、H6牧场ROH比例较大(83.70%、84.30%),其余牧场ROH比例范围为73.33%~76.52%;在5~10 Mb长度上,ROH总体比例占15.14%,其中H1、H6牧场ROH比例较小(10.26%、10.67%),其余牧场ROH比例范围为14.89%~17.06%;在>10 Mb长度上,ROH总体比例占8.64%,其中H1、H6牧场ROH比例较小(6.03%,5.04%),其余牧场ROH比例范围为7.61%~9.61%。图2展示了常染色体上不同长度ROH的数目。

图2 染色体上不同长度ROH的数目Fig.2 Number of ROH with different length on chromosome
2.3 基因组近交系数评估
不同牧场荷斯坦牛群体基于ROH的近交系数及变化范围见表2。全群中基于ROH的基因组FROH范围为0.021~0.447,近交系数平均值为0.106,标准差为0.040。其中H2号牧场平均FROH最高(0.123),H7号牧场平均FROH最低(0.082),其他牧场分别为0.112、0.114、0.109、0.108、0.103。在个体层面中,FROH最低的个体出现在H7号牛场中(0.021),FROH最高的个体出现在H4号牛场中(0.447)。
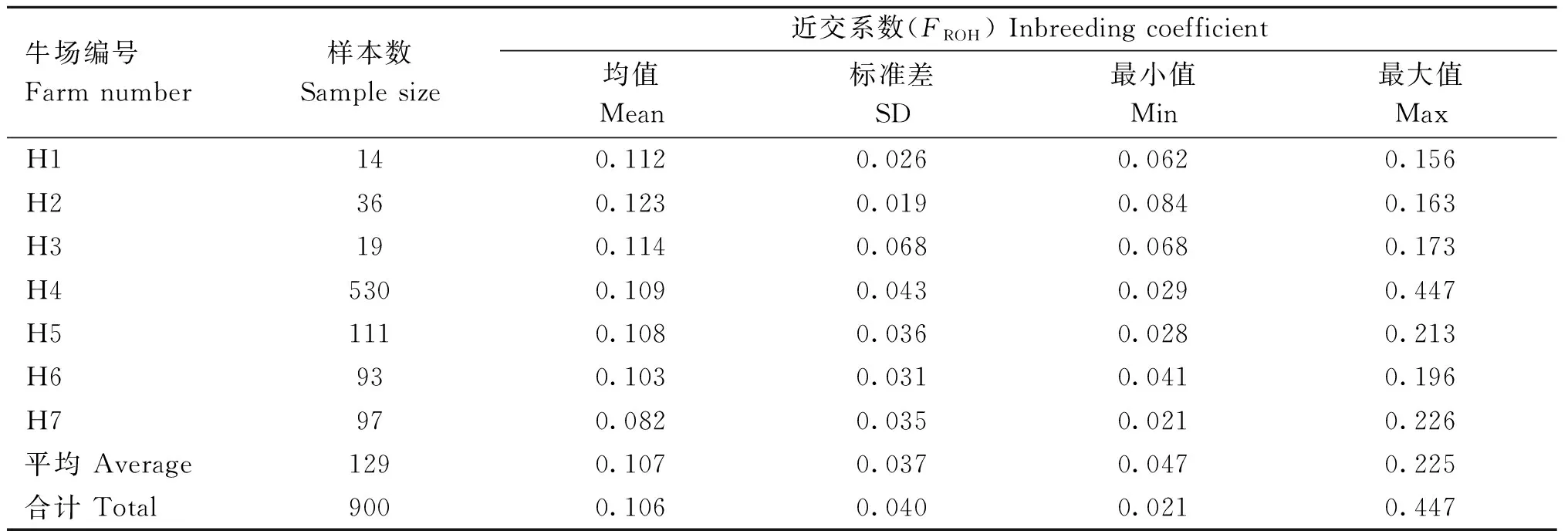
表2 基于ROH的不同奶牛场的近交系数(FROH)Table 2 Inbreeding coefficient (FROH) of different dairy farms based on ROH
2.4 高频ROH区域及候选基因鉴定与注释、富集
图3展示了在1~29号染色体上组成ROH的SNPs占群体的百分率。通过选择组成ROH中前1% SNPs,以确定统计阈值,本研究选取频率大于29.78%作为高频率的ROH区域阈值。共检测到8个高频区域,并通过Ensembl数据库对ROH中的高频区域进行基因注释,共注释到79个基因,见表3。其中,14号染色体上22.78~23.38 Mb位置的区域,80%的个体都在该区域内发生ROH片段,并注释到3个基因。利用KOBAS对注释到的基因进行KEGG通路富集分析,结果见表4。分析得出79个基因显著富集于酮体的合成与降解(synthesis and degradation of ketone bodies)、缬氨酸、亮氨酸和异亮氨酸降解(valine, leucine and isoleucine degradation)、丁酸代谢(butanoate metabolism)、Ras信号通路(ras signaling pathway)等11个信号通路。

表3 荷斯坦牛高频ROH区域及候选基因Table 3 High-frequency ROH regions and candidate genes in Holstein cattle

表4 高频ROH区域基因的KEGG通路富集分析(P<0.05)Table 4 KEGG pathway enrichment analysis of genes in high-frequency ROH regions(P<0.05)

图3 ROHs中SNPs百分比曼哈顿图Fig.3 Manhattan plot of SNPs percentages in ROHs
3 讨 论
3.1 荷斯坦牛群体基因组ROH基本统计分析
不同育种目标及选择强度会引起不同荷斯坦牛群体中ROH数目、长度及分布情况的差异[5-6,27]。Kim等[7]通过比较3个北美荷斯坦牛群体在产奶性状不同选择强度下基因组ROH的变化,揭示了总体ROH频率和分布方面的显著差异,结果显示群体内ROH平均长度约为6 Mb,小于5 Mb的ROH片段数目占总片段数目的53%。而与Kim等[7]的研究结果相比,本研究中荷斯坦牛群体ROH平均长度为4.23 Mb,小于5 Mb的ROH片段的数目占总片段数目的76.21%。另外对比不同牧场群体,小于5 Mb的ROH片段数目所占比例也有差异。在基因组ROH长度上,Marras等[28]利用50K SNP芯片对5个意大利公牛品种进行ROH分析,结果表明相较于其他品种,乳用品种荷斯坦牛和意大利布朗牛的平均ROH长度更大(3.6、3.9 Mb),其中荷斯坦牛群体的ROH平均长度与本研究的结果相近。在牧场群体方面,H1和H6号牧场群体在小于5 Mb的ROH片段数目占总片段数目最高(83.70%、84.30%),而大于10 Mb的ROH片段数目占总片段数目比例最低(6.03%、5.04%)。研究显示,较近世代的共同祖先会造成长ROH片段的形成,短的ROH来源于关系较远的共同祖先[7,29]。此外,各个牧场奶牛群体ROH平均长度、变化范围也有差异,这与不同牧场奶牛群体来源以及选配过程中使用不同国别的冷冻精液有关。因此,本研究基于对不同牧场群体基因组ROH的数目、长度及分布的研究,评估群体近交情况,为牧场今后的选种选配提供参考。
3.2 基于ROH的基因组近交系数
目前,ROH常用来计算个体近交系数,且具有较高的准确性[15,30-33]。本研究中,河南荷斯坦牛群体总平均FROH(0.106)与宁夏[21](0.101)、北京[14](0.007~0.312)荷斯坦牛群体FROH相近,与上海[17]荷斯坦牛群体(0.363)相差较大。上海与北京作为我国的南、北奶牛养殖业的代表地区,由于选育目标、强度、气候等因素的影响,群体近交程度出现差异,河南地理位置上属于中原地区,在奶牛育种策略和群体近交情况上与北方更相近。近交水平在一定程度上也可以反映牧场选种选配管理状况。在牧场选配管理上,由表2可以看到,H1、H2、H3号牧场平均FROH较高(0.112、0.123、0.114),H7号牧场平均FROH较低(0.082),不同牧场之间的差异侧面反映出这些牧场在选配过程中对群体近交问题的管理程度;在牧场规模上,H1、H2、H3号牧场规模较小,群体数量较少,平均FROH较高(0.112、0.123、0.114),H4号牧场规模较大,群体数量多,平均FROH较低(0.109)。此外,在H4号牧场中有些个体的FROH明显较高(>0.285),最大FROH达到0.458,反映出该牧场在个体选种选配过程中未充分考虑近交问题。因此,通过对近交系数的计算可以了解不同牧场群体近交状况,从而在实际选种选配工作中能更有效的避免近交,减少经济损失。
3.3 基因组高频ROH区域的候选基因分析
本研究在高频ROH区域中共鉴定到了79个基因,其中包含与奶牛经济性状有关的基因,如AKAP3、C5H12orf4、CAPN3、ARL15、XKR4、CRBN、IL5RA等。5号染色体上AKAP3、C5H12orf4、FGF6基因与体型、体高有关[34-36]。10号染色体上CAPN3基因与胴体、繁殖性状相关[37-38]。CHST14基因与妊娠维持和胎儿生长直接相关[39]。22号染色体上IL5RA基因影响牛奶蛋白质组成[40]。此外还有一些基因与繁殖、生长等性状有关,如FGF10基因参与调节胎儿卵泡生成[41]。值得注意的是,14号染色体上22.78~23.38 Mb区域是ROH频率最高的区域,80%的个体都在该区域内发生ROH片段(图3)。发现该区域与宁夏[21]荷斯坦牛群体高频区域(21.61~24.99 Mb)高度重合,这可能与不同地区育种目标及选择强度有关,并随着选育的推进,在基因组中出现相近的长纯合区域。这个高频区域注释到TGS1、LYN、CHCHD7基因,这些基因与生长、胴体相关性状[42-43]和饲料效率有关[35,44-45]。因此,本研究在ROH富集区域鉴定的基因可以为荷斯坦奶牛分子育种提供候选基因信息。
4 结 论
本研究对河南省荷斯坦牛群体进行全基因组ROH检测与分析,发现ROH在不同牧场群体中的数目、长度及频率存在差异,基于ROH计算的近交系数范围在0.082~0.123,反映出不同牧场近交水平存在差异,这有助于了解河南省荷斯坦牛群体近交程度,为牧场选育过程中避免近交提供指导。在全基因组范围内检测到8个高频ROH富集区域,共筛选出79个与奶牛经济性状相关的基因,如AKAP3、C5H12orf4、CAPN3、ARL15、XKR4、CRBN、IL5RA等,可作为奶牛分子育种中进行标记辅助选择的候选基因。
猜你喜欢
小猕猴智力画刊(2021年6期)2021-08-05
海峡姐妹(2020年6期)2020-07-25
中国民族医药杂志(2016年5期)2016-05-09
作文大王·低年级(2016年3期)2016-03-11
儿童故事画报·智力大王(2015年12期)2016-01-23
华南农业大学学报(2015年5期)2015-12-04
畜牧兽医学报(2015年3期)2015-07-05
小学生时代·大嘴英语(2014年5期)2014-11-05
云南畜牧兽医(2014年2期)2014-02-28
当代畜禽养殖业(2014年6期)2014-02-27
