
Hepatic ischemia-reperfusion syndrome and its effect on the cardiovascular system: The role of treprostinil, a synthetic prostacyclin analog
2023-10-21 01:03ChristinaMouratidouEfstathiosPavlidisGeorgiosKatsanosSerafeimChrysovalantisKotoulasEleniMouloudiGeorgiosTsoulfasIoannisGalanisTheodorosPavlidis
Christina Mouratidou, Efstathios T Pavlidis, Georgios Katsanos, Serafeim-Chrysovalantis Kotoulas, Eleni Mouloudi, Georgios Tsoulfas, Ioannis N Galanis, Theodoros E Pavlidis
Abstract Hepatic ischemia-reperfusion syndrome has been the subject of intensive study and experimentation in recent decades since it is responsible for the outcome of several clinical entities, such as major hepatic resections and liver transplantation. In addition to the organ’s post reperfusion injury, this syndrome appears to play a central role in the dysfunction of distant tissues and systems. Thus, continuous research should be directed toward finding effective therapeutic options to improve the outcome and reduce the postoperative morbidity and mortality rates. Treprostinil is a synthetic analog of prostaglandin I2, and its experimental administration has shown encouraging results. It has already been approved by the Food and Drug Administration in the United States for pulmonary arterial hypertension and has been used in liver transplantation, where preliminary encouraging results showed its safety and feasibility by using continuous intravenous administration at a dose of 5 ng/kg/min. Treprostinil improves renal and hepatic function, diminishes hepatic oxidative stress and lipid peroxidation, reduces hepatictoll-like receptor 9 and inflammation, inhibits hepatic apoptosis and restores hepatic adenosine triphosphate (ATP) levels and ATP synthases, which is necessary for functional maintenance of mitochondria. Treprostinil exhibits vasodilatory properties and antiplatelet activity and regulates proinflammatory cytokines; therefore, it can potentially minimize ischemia-reperfusion injury. Additionally, it may have beneficial effects on cardiovascular parameters, and much current research interest is concentrated on this compound.
Key Words: Hepatic ischemia-reperfusion syndrome; Myocardial damage; Prostaglandins; Treprostinil; Liver transplantation; Hepatectomy
INTRODUCTION
Hepatic ischemia-reperfusion syndrome has been the subject of intense study and experimentation in recent decades since it is responsible for the outcome of several clinical events, such as hemorrhagic shock, major hepatic resections, Budd-Chiari syndrome and some types of hepatotoxicity[1-3]. Worldwide, end-stage liver disease is a common cause of morbidity and mortality, and liver transplantation remains the gold standard therapy for these patients. Nevertheless, prolonged exposure of the graft to cold and warm ischemia has a direct risk of serious postoperative complications, such as poor early graft function and primary nonfunction[4,5].
The complex blood supply in combination with the increased metabolic activity of the liver and its involvement in homeostasis, detoxification, protein synthesis, energy storage and immunity processes render the organ extremely sensitive to circulatory disorders. Liver ischemia-reperfusion syndrome remains a major cause of worse postoperative clinical outcomes. The pathophysiological changes do not pertain to single organ damage but also to a complex systemic process that affects other structures and tissues, causing a cascade of multiple organ dysfunction[6-9].
The responsible mechanisms are exceedingly complicated and involve numerous factors, including mediators, cytokines, adhesion molecules, vasoactive agents and reactive oxygen species. During an ischemic period, several functional processes take place at the cellular level and stimulate cell injury[9,10]. The exposure of hepatocytes to low oxygen levels results in changes in intracellular pH and a decrease in adenosine triphosphate (ATP) production, thereby attenuating the intrahepatic energy content[11]. Excessive production of reactive oxygen species and reactive nitrogen species in mitochondria and intracellular calcium overload promote organelle destruction and cell death. Innate immunological processes involve the activation of liver Kupffer cells; the accumulation of circulating lymphocytes, neutrophils, platelets and monocytes; and hepatic macrophage polarization and differentiation[12-14]. In this respect, Kupffer cells produce reactive oxygen species, interleukin (IL)-1, and tumor necrosis factor (TNF)-α, thereby triggering the recruitment of CD4+ T lymphocytes. In turn, activated CD4+ T cells can trigger Kupffer cells, leading to aggravation of the inflammatory response. Concurrently, Kupffer cells may have a protective role by producing anti-inflammatory IL-10 and suppressing the expression of proinflammatory factors, such as TNF-α, IL-1β, interferon-γ, and IL-2, and adhesion molecules, such as intercellular adhesion molecule 1[15,16]. Disrupted liver metabolism elicits an endogenous inflammatory cascade, which includes excessive cytokine and chemokine production, the release of adhesive molecules and caspase-1 activation[17,18]. Blood flow restoration and re-exposure of ischemic hepatocytes to high oxygen level conditions contributes to further hepatocellular damage, mediated by reactive oxygen species generation[19]. An inflammatory outbreak of hepatic ischemia-reperfusion has been found to initiate a series of pleiotropic mitogen-activated protein kinase (MAPK) cascades. Among them, the activated P38 and c-Jun N-terminal kinase (JNK) cascades are most involved in the pathways of apoptotic or autophagic hepatic cell death[20,21]. Notably, current evidence suggests that the MAPK, mammalian target of rapamycin and nuclear factor kappa B (NF-κB) inflammatory signals are adjusted by tripartite motif containing protein 37, which plays important role in exacerbation of hepatic ischemia-reperfusion injury by directly interacting with TNF receptor-associated factor 6 (TRAF6)[22]. Cellular damage has been shown to be promoted by the downregulation of microRNAs (miRNAs), which are small, single-stranded, noncoding RNA molecules. Specifically, suppression of miRNA-142-3p, miRNA-146a, miRNA-200c, and miRNA-34a is suggested to worsen the condition of hepatic ischemia-reperfusion injury, while the inhibition of miR-450b-5p has the opposite response[23,24]. On the other hand, miR-125b attenuates hepatic ischemia-reperfusion injury by suppressing TRAF6 and NF-κB signal pathways[25]. In general, long non-coding RNA and miRNAs regulatory networks mediate the pathological progression of hepatic ischemia-reperfusion injury through mutual activation and interference[26].
Hepatic ischemia-reperfusion syndrome mechanisms and its systemic effects
Hepatic ischemia-reperfusion syndrome is associated with several vascular disorders, such as increased vascular permeability, endothelial cell edema and loss of homeostasis between vasoconstricting and vasodilating factors. Accumulated neutrophils form neutrophil extracellular traps (NETs) that have been shown to play a significant role in the interactions with platelets and are involved in pro-coagulation mechanisms in a variety of infectious and sterile inflammatory processes. A recent study demonstrated that hepatic ischemia-reperfusion leads to a NET-mediated hypercoagulable state and subsequent organ injury through microvascular immuno thrombi formation[27]. Further liver microcirculatory milieu obstruction results in deterioration of ischemic hepatocellular damage and cell death. Although hepatic cell injury appears to progress primarilyviathe lytic necrosis pathway, it seems that more complex, often complementary or overlapping mechanisms of programmed cell death occur based on the presence or absence of damageassociated molecular patterns (DAMPs). These mechanisms can be categorized into inflammatory, such as necrosis, necroptosis, pyroptosis, and ferroptosis, and noninflammatory subtypes, such as apoptosis[13,28-30]. In conclusion, the hepatic ischemia-reperfusion mechanisms are summarized in Table 1.
Severe hepatic ischemia-reperfusion injury does not constitute only a local phenomenon. It is characterized by a widespread systemic sterile inflammatory response with the accumulation of inflammatory cells in distant organs. Reactive oxygen radicals that are released following ischemic hepatocyte reperfusion promote systemic oxidative stress, resulting in remote organ damage[31-35]. In addition, platelet aggregation induces a procoagulant state and associated ubiquitous platelet-rich microvascular thrombus formation. Systemic NET-mediated hypercoagulability leads to remote organ injury through platelet toll-like receptor 4 (TLR4)-dependent signaling pathways[36]. These underlying mechanisms are responsible for the dysfunction of other organs, including the lung, kidney, intestine, pancreas, brain, and myocardium (as shown in Figure 1)[1,37,38]. The resulting multiple organ dysfunction syndrome occurs as a progressive, complex and dynamic process with a variable extent of organ failure and a direct deteriorating effect on survivorship[39].
Effect of hepatic ischemia-reperfusion syndrome on the cardiovascular system
Multiple organ dysfunction is a major complication of acute liver failure. The incidence of this particularly severe condition is approximately 1-8 cases per million inhabitants, and it is responsible for 6% of deaths due to liver disease and up to 7%-8% of liver transplants[40]. Although there are several reports of acute liver failure cases followed by myocardial involvement, the direct effects of hepatic ischemia-reperfusion syndrome on the myocardium have not been analyzed completely. Significantly elevated cardiac troponin I and creatine phosphokinase myocardial band (CK-MB) values have been associated with increased mortality, while the incidence of major cardiovascular events is undoubtedly higher in patients with acute liver failure[1,30,41]. Troponin I is a sensitive and myocardium-selective biomarker with both prognostic and diagnostic value. Troponin has become ingrained in the Universal Definition of Acute Myocardial Infarction but may also be detected in stable chronic conditions[42]. However, there is a high prevalence of elevated troponin in noncardiac clinical conditions, such as myocarditis, pulmonary embolism, acute heart failure, septic shock, and drug-induced cardiotoxicity, as well as after interventional procedures such as coronary angioplasty and electrical cardioversions. Thus, measurement of troponin elevation, especially with high-sensitivity assays, allows detection of clinical cases with nonacute coronary syndrome-mediated myocardial injury[43,44]. CK-MB is also preferred in particular situations, specifically in the diagnosis of acute myocardial infarction and cardiac injury evaluation. Although it has limitations in terms of early diagnosis, elevated CK-MB levels reveal myocardial damage secondary to some noncardiac conditions[45,46].
Clinical and pathophysiological variability in the remote organ impairment following acute liver failure is a result of the complicated interactions. Mitochondrial dysfunction and impaired ATP production are characteristic features and lead to energy balance disruption. As a consequence, parenchymal cells are forced to alter their metabolic activity to maintain their energy provision by enhancing proteolysis and lipolysis[47]. Pathogen-associated molecular patterns (PAMPs) and DAMPs released by damaged cells reinforce the systemic immune response and trigger cell death[48,49].
Severe circulatory disturbances are also observed in patients with acute liver failure, regardless of the cause of liver disease. Hyperdynamic circulation is characterized by markedly elevated cardiac output and low systemic vascular resistance due to peripheral vasodilation. These pathophysiological cardiovascular changes are similar to those seen in patients with septic shock[50]. Changes in the microcirculation during acute liver failure have also been described. Intrahepatic and systemic microcirculation abnormalities include vasoconstriction, precapillary shunt formation and reduced blood flow resulting in loss of multiorgan function[51].
In some experimental models, increased cardiac enzyme levels and histopathological myocardial tissue damage were not attributed only to metabolic stress and hemodynamic instability. Other complex mechanisms, such as inflammation, endothelial cell disorders and the production of reactive oxygen and nitrogen species, were also observed[41,47]. The histological examination of animal heart tissue demonstrated wavy fibers, which are consistent with myocardial infarction and the presence of microthrombi in the capillary area of the myocardium, whereas the perivascular lesions were rather unrepresentative, supporting the idea of a mechanism of injury originating from the vascular system[41].
The postischemic phase is characterized by liver parenchymal dysfunction and the secretion of proinflammatory cytokines. Excessive TNF-α and IL-6 production and the systemic inflammatory response contribute to distant organ damage. Reactive oxygen species and cytokines generated during the reperfusion phase flow from the hepatic veins directly to the right atrium. Thus, the heart is the first organ receiving blood flow from postischemic hepatic tissue, which makes it more susceptible to damage[52,53]. The reactive oxygen radicals generated at the onset of reperfusion result in both direct cellular damage (necrosis, membrane disruptions) and indirect damage through cellular signaling pathways[54,55]. In a recent study, a histopathological heart examination of animals subjected to hepatic ischemia-reperfusion demonstrated necrosis, hyperemia, hemorrhage, and edema of myocardial cells[52].
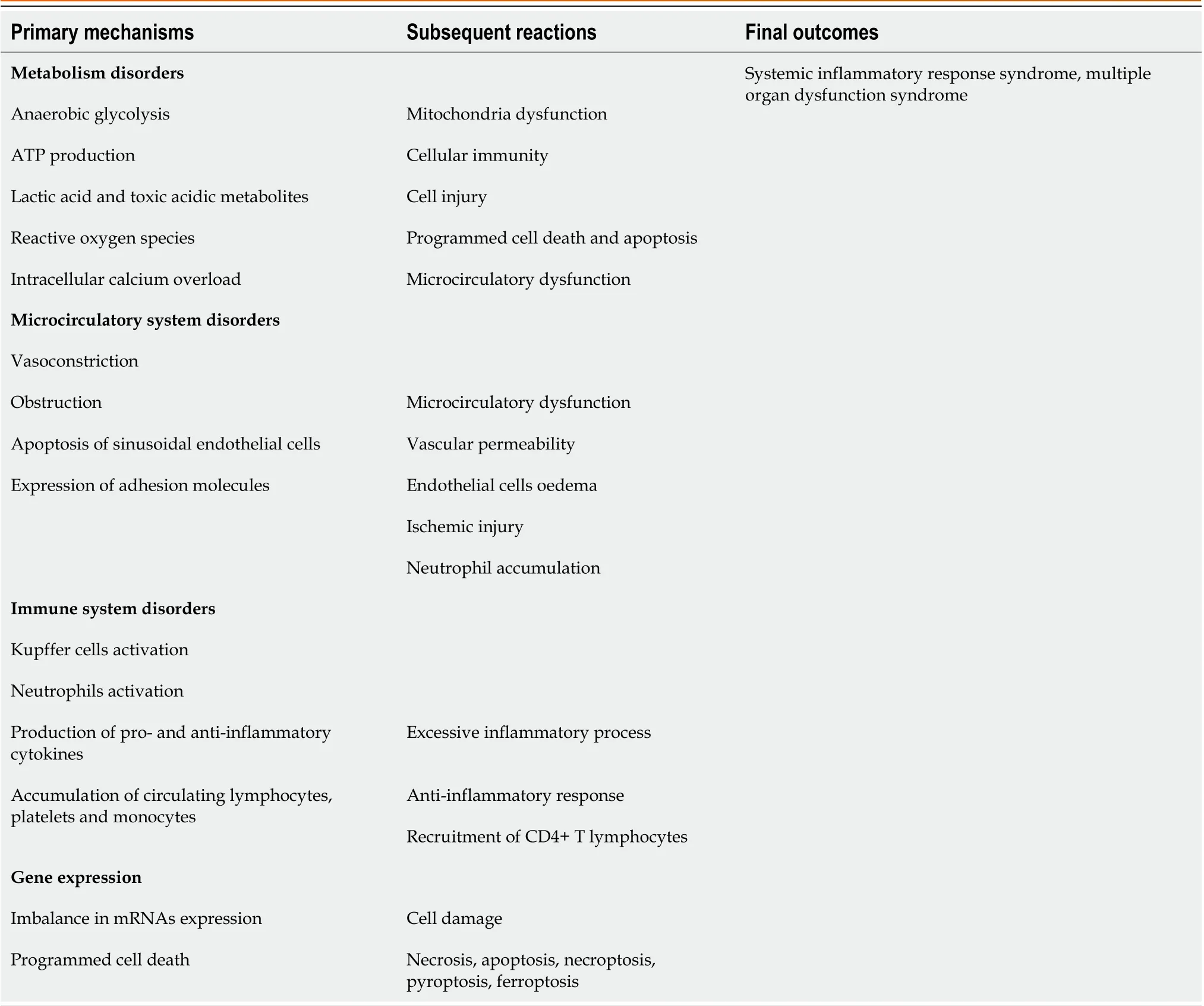
Table 1 Hepatic ischemia-reperfusion injury mechanisms
Calcineurin is a calcium- and calmodulin-dependent serine/threonine protein phosphatase that plays an important role in T-cell activation, transcription regulation, cell cycle control and apoptosis[56]. In the heart, calcineurin is primarily present in the context of the hypertrophic growth response and pathological cardiac remodeling due to its role in nuclear factor of activated T cells transcription factor activation[57,58]. The inhibition of the calcineurin signaling pathway by tacrolimus attenuates myocardial damage after total hepatic ischemia-reperfusion. Furthermore, the protective role of tacrolimus in stabilizing the mitochondrial membrane potential, avoiding impairment in mitochondrial respiration and oxidative phosphorylation, improving antioxidant capacity, and reducing calcium overload prevent the myocardium from experiencing cell injury and potentially cell death[59,60]. In general, regulation of calcium homeostasis showed effectiveness on protecting hepatocytes from ischemia-reperfusion injury, such as protection during cardiac arrhythmias. A recently discovered HBF001 heparin fragment acts on sodium-calcium exchanger, by altering peptide structure and accelerating the intracellular calcium output[61].
Hepatic ischemia may induce a series of biochemical reactions, including modifications in the interactions between factors controlling programmed cell death and apoptosis. In a recent experimental study, increased levels of the proapoptotic protein Bax and decreased levels of the antiapoptotic protein Bcl-2 were measured. According to the article, hepatic ischemia-reperfusion injury accelerated apoptosis of myocardial cells and damaged the myocardium. Likewise, based on cardiac function observations, the ventricles of animals were enlarged and thickened, and ventricular systolic function was decreased in the control group[62-64].
Occlusion of the hepatic artery and the portal vein may be necessary to avoid excessive bleeding during major hepatectomy and liver transplantation. However, total hepatic vascular exclusion is associated with profound volume shifts due to preload reduction, resulting in a decrease in cardiac output and hemodynamic instability[65,66]. Chenet al[67] showed that decreased left ventricular preload was the primary reason for the reduced cardiac output, stroke volume and ejection fraction during liver ischemia. Along with impaired cardiovascular function, the systemic inflammatory response and activated neutrophil accumulation in the myocardium are simultaneously responsible for remote organ injury induced by hepatic ischemia-reperfusion[56,57].
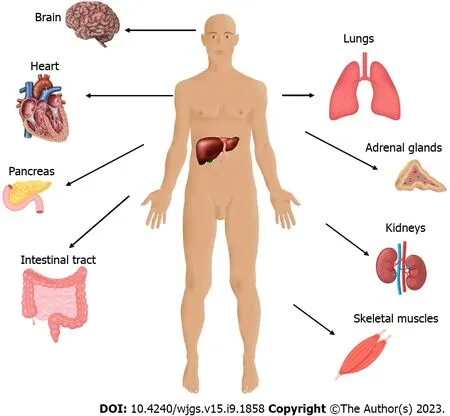
Figure 1 Remote organ damage after hepatic ischemia-reperfusion injury.
Myocardial injury appears to be a serious complication of hepatic ischemia-reperfusion syndrome, but the clinical manifestations in humans have not yet been established. Only a small percentage of patients who underwent liver transplantation and presented with mild hepatic ischemia-reperfusion injury (aspartate aminotransferase < 2000 IU/L) developed severe impairment of the left ventricular ejection fraction (< 35%)[68]. Despite being uncommon, post liver transplantation cardiac dysfunction remains a major clinical problem. Although the evidence supporting the idea of a direct association between hepatic ischemia-reperfusion and deterioration of left ventricular systolic function is still inconclusive, it is possible that the systemic inflammatory response and hemodynamic instability can contribute to postoperative cardiomyopathy[69]. Several studies have described an incidence of acute posttransplantation systolic heart failure of 1%-7%, frequently caused by stress-induced cardiomyopathy. However, hepatic ischemia-reperfusion syndrome was not exactly identified as an etiological underlying condition[70,71]. Intraoperative cardiovascular stress negatively affects preexisting cardiac dysfunction. Theoretically, hepatic ischemia-reperfusion syndrome presenting in the surgical postreperfusion phase may be associated with myocardial depression, pulmonary arterial hypertension, a significant reduction in systemic vascular resistance and bradycardia. Reactive oxygen species and multiple inflammatory mediators, such as cytokines and chemokines, are also responsible for the clinical phenotype of postreperfusion syndrome[72-74].
Although the link between hepatic ischemia-reperfusion syndrome and myocardial injury has been made in animal models, the consequences of this effect have yet to be defined. Myocardial damage has been described mostly as histopathological lesions and altered laboratory findings rather than serious clinical manifestations. After all, it must not be forgotten that most experimental and clinical observations are made in relatively healthy individuals and not in those whose heart is already affected by ischemia, cardiomyopathy and systolic/diastolic dysfunction[1,75,76]. Apparently, cardiac impairment following hepatic ischemia-reperfusion syndrome encompasses a large spectrum of subclinical and symptomatic conditions, which are responsible for additional short- and long-term morbidity and low survival[74]. Thus, continuous research should be directed to finding effective therapeutic options to improve the outcome and reduce the postoperative mortality rates.
The therapeutic use of prostaglandins
The therapeutic strategies against hepatic ischemia-reperfusion syndrome mainly include protective intraoperative techniques and an adequate number of pharmacological agents. Prostaglandins are a group of physiologically active lipid compounds called eicosanoids, which consist of oxidized derivatives of 20-carbon polyunsaturated fatty acids, primarily arachidonic acid, formed due to the cyclooxygenase pathway[77-79]. Many studies have shown the liver cytoprotective ability of prostaglandins based on direct or indirect signaling pathways[80]. The effectiveness of prostaglandin analogs has been evaluated in several experimental models along with patients who underwent liver transplantation. Prostaglandin E1 (PGE1) administration has been shown to improve liver microcirculation dysfunction by the expansion of blood vessels and enhancement of perfusion status. PGE1 also downregulates the expression of adhesion molecules and inflammatory mediators, resulting in inhibition of platelet aggregation and leukocyte adherence. Furthermore, suppression of thromboxane A2 in combination with the reduction in protease release and oxygen free radical production leads to attenuation of the inflammatory cascade and minimization of the sinusoidal cell apoptosis rate[81-84]. Prostacyclin (PGI2) is another member of the prostaglandin family with potential vasodilating, antithrombotic and anti-inflammatory effects. Prostacyclin analogs have been established for decades in the treatment of patients with pulmonary arterial hypertension and remain an integral component of the current therapeutic armamentarium[85-87]. Preconditioning with beraprost sodium, a prostacyclin analog, in the experimental hepatic ischemia-reperfusion model led to suppressed production of the inflammatory mediators TNF-α and IL-1β, and attenuation of hepatic cell apoptosis in a dose-dependent manner. Through inhibition of the phosphorylation of P38 and JNK signaling cascades, beraprost sodium could ameliorate the systemic inflammatory response, apoptosis and autophagy processes of hepatic ischemiareperfusion[21]. Although several studies have shown beneficial effects of prostaglandin therapy in the prevention of liver damage following transplantation, the clinical utility of these agents is rather limited due to their unstable structure, serious adverse reactions and very short half-lives[77,82,88,89].
The protective role of treprostinil
Treprostinil is a relatively new prostaglandin I2 (PGI2, prostacyclin) analog with a stable structure (as shown in Figure 2), longer half-life and improved potency that has been approved by the Food and Drug Administration (FDA) in the United States since 2002 for the treatment of patients with pulmonary arterial hypertension[86]. Treprostinil demonstrated stability for 48 h at 40 °C in different solutions and multiple beneficial effects, promoting its administration in long-term therapy[90-93]. Its binding profile and corresponding biochemical cellular response on human prostanoid receptors have been sufficiently analyzed. Treprostinil has high affinity for the DP1, EP2and IP receptors; low affinity for EP1and EP4receptors; and even lower affinity for EP3, FP and TP receptors[94]. The mechanisms of action of treprostinil are summarized in Table 2. In general, the PGI2 signaling pathways are much more complex than anticipated and remain incompletely elucidated. Peroxisome proliferator-activated receptors constitute an important signaling pathway that partially explains the vasodilating effect of prostacyclin, along with its cytoprotective properties[95,96]. The effects of treprostinil on angiogenesis have also been reported, including the vascular endothelial growth factor (VEGF)/NADPH oxidase 4 signaling pathway.In vitrotreprostinil administration enhanced VEGF-A synthesis by mesenchymal stem cells, resulting in activation of vessel-forming ability[97].
Experimental orthotopic liver transplantation in rats with subcutaneous treprostinil administration at a dose of 100 ng/kg/min showed very encouraging results[98]. Specifically, treprostinil increased liver blood flow during the reperfusion phase while supporting the balance within the vasculature by increasing intracellular cyclic adenosine monophosphate (cAMP) levels. Furthermore, inhibition of platelet aggregation and proinflammatory cytokine production in the early posttransplantation period protected the liver graft against hepatic ischemia-reperfusion injury. Additionally, Houet al[99] demonstrated that treprostinil improves renal and hepatic function, diminishes hepatic oxidative stress and lipid peroxidation and reduces hepatic TLR9, which is located in endosomes and triggers the inflammatory response by recognizing PAMPs and DAMPs[100]. Another recent study suggested that the presence of Gs-coupled prostanoid receptors in liver sinusoidal endothelial cells was responsible for the beneficial effect of prostaglandins. In fact, treprostinil binds and activates EP2, EP4, and IP receptors, resulting in attenuation of ischemia-induced hepatic cell injury[101].
Mitochondrial dysfunction during hepatic ischemia-reperfusion leads to increased DNA fragmentation and induction of programmed cell apoptosis. To maintain mitochondrial homeostasis and mediate acute cell injury, a complex fundamental process, named mitochondrial biogenesis, typically occurs in response to postischemic cellular stress. Induction of mitochondrial biogenesis is mediated by upregulation of the transcription factor peroxisome proliferatoractivated receptor gamma coactivator 1-alpha (PGC-1α), which is considered to be the master regulator for the process and has been found to be significantly decreased in ischemia-reperfusion injury[99,102-104]. Treprostinil upregulatesPgc-1αmRNA expression, thus securing mitochondrial biogenesis and improving mitochondrial dynamics. Additionally, treprostinil inhibits hepatic apoptosis by suppressing the release of mitochondrial cytochrome c and caspase-3 activation. In general, treprostinil restores ATP production, which ameliorates hepatic mitochondrial injury and preserves cellular energy balance[99,105,106].
Several studies have shown that hepatic ischemia-reperfusion injury may cause a reduction in hepatic cytochrome P450 (CYP) levels and/or changes in enzyme activity amplitude. CYP has a broad range of functions, including drug metabolism and clearance and detoxification of pharmaceutical substances. The excessive cytokine release and systemic inflammatory response during ischemia-reperfusion injury have been associated with reduced microsomal drug metabolism, which can cause dose-dependent drug toxicity[107-109]. Treprostinil administration improvedCYPmRNA expression in liver grafts after clinically relevant rat liver transplantation. In addition, treprostinil restored CYP protein expression and improved its activity in liver grafts[110]. The results showed that extended hepatic ischemia-reperfusion injury impaired CYP450 protein expression for at least 48 h post-transplantation, while treprostinil administration improved the protein expression of the three major CYP450 enzymes (CYP3A2, CYP2C11, and CYP2E1) in the liver graft and promoted CYP450-mediated drug metabolism[110].
Isolated rat liver perfusion is a widely performedex vivoexperimental model and represents a suitable tool for studying various pathological conditions, such as hepatic ischemia-reperfusion injury[111]. A recent study performed on isolated rat livers demonstrated the effect of postischemic hepatic injury on the expression of basolateral (uptake) and apical (efflux) hepatic drug transporters, which was significantly altered[112]. Importantly, treprostinil administration at a dose of 20 ng/mL during preservation and/or reperfusion reduced the ischemia-reperfusion-mediated effects on the expression of the Slc10a1/Ntcp and Slc22a1/Oct1 drug uptake transporters, similar to the expression of the apical efflux drug transporter P-gp (Mdr1a, Abcb1a). Although these findings illustrated improved liver function due to treprostinil supplementation, deeper knowledge is needed to determine the effect of the particular synthetic prostacyclin on the expression of drug-metabolizing enzymes and the regulation of drug transporters[113].

Table 2 Treprostinil mechanisms of action

Figure 2 The chemical structure of prostaglandin I2 and prostacyclin analogue treprostinil.
Liver graft injury post-transplantation frequently presents with elevated bilirubin and amino-transaminase serum levels during the first 24 h following transplantation. Many studies support the hypothesis of hepatic ischemiareperfusion syndrome as the leading cause of initial poor graft dysfunction and primary graft nonfunction[114-117]. However, no pharmacological options are currently approved for the prevention of hepatic ischemia-reperfusion injury following transplantation. A prospective, pilot, single-center, open-label, nonrandomized, dose-escalation phase I/II study in liver transplant patients investigated the efficacy of intravenous treprostinil administration in the prevention of hepatic ischemia-reperfusion with some encouraging results[118]. A small group of patients who underwent liver transplantation and received perioperative intravenous treprostinil at a dose of 5 ng/kg/min followed by postoperative contentious infusion at a dose of 2.5-5 ng/kg/min for approximately 5 d showed improved liver function and 100% graft and recipient survival at six months[118]. Preliminary observations indicated a rapid reduction in transaminase plasma levels, improvement in hepatobiliary excretory function and prevention of the occurrence of acute kidney failure. Furthermore, stable hemodynamic parameters in the patients with treprostinil administration during the study period were achieved, since the mean pulmonary arterial pressure, systemic blood pressure, and cardiac index values remained within the normal range.
The initial phase of hepatic injury is characterized by ATP depletion, mitochondrial dysfunction and reactive oxygen species accumulation, followed by a systemic sterile inflammatory response. In general, oxidative and inflammatory pathways have been shown to play an important role in remote organ functional changes in a state of hepatic ischemiareperfusion injury. Although myocardial impairment is documented mostly as a subclinical event, the general clinical status of remote organ damage in the postreperfusion phase can directly affect overall survival rates. Additionally, while a hypothesis of myocardial injury in the setting of hepatic ischemia-reperfusion has already been reported, the consequences of the particular issue remain unclear[116-118].
Over the last few years, treprostinil has become one of the key therapeutic options for the treatment of patients with pulmonary arterial hypertension[119]. Along with its beneficial influence on pulmonary vascular smooth muscle proliferation, vasoconstriction and pulmonary vascular remodeling, treprostinil also shows a direct favorable effect on cardiac function[120,121]. Experimental treprostinil administration increased stroke volume and cardiac output, leading to a stable hemodynamic state and improved cardiovascular endurance[121-123]. In addition, a broad reduction in reactive oxygen species accumulation and lipid peroxidation and a decrease in cytokine and chemokine mRNA levels during ischemia-reperfusion may protect the myocardium from postreperfusion injury. However, there are several reports of beneficial effects of prostacyclin analogs on the attenuation of myocardial ischemia-reperfusion injuryviavasodilation, inhibition of platelet accumulation and anti-inflammation[124,125]. Finally, the acceleration of mitochondrial recovery due to reduced mitochondrial-mediated cell apoptosis supports the hypothesis of treprostinil-mediated organ protection against ischemia-reperfusion injury[104]. Although currently available data are not sufficient, there are several indications of the beneficial effect of treprostinil on remote organ damage in the course of hepatic ischemia-reperfusion syndrome. Recent studies with subcutaneous treprostinil administration in experimental hepatic ischemia-reperfusion models have shown very encouraging results. Furthermore, patients with pulmonary arterial hypertension treated with treprostinil demonstrated an improved hemodynamic state and stable cardiac parameters[123].
CONCLUSION
Hepatic ischemia-reperfusion syndrome is a major complication of liver surgery, including partial liver resection and liver transplantation, liver trauma, resuscitation and other clinical entities. The pathophysiological mechanisms of hepatic ischemia-reperfusion are not responsible for liver damage alone but also occur as a complex systemic process with a direct impact on the function of multiple tissues and organs. Moreover, in some cases, postreperfusion systemic injury can lead to systemic inflammatory response syndrome and/or multiorgan dysfunction syndrome, both of which have a high incidence of mortality and morbidity. Thus, therapeutic strategies, including advanced surgical techniques and pharmacological inhibitors, should be studied intensively to improve the outcome of these patients. Treprostinil is a relatively new, FDA-approved stable prostacyclin analog with potent anti-inflammatory, antifibrotic, vasodilating, antiremodeling and antiapoptotic activities. According to current knowledge, there is a positive correlation between treprostinil supplementation and the attenuation of liver ischemia-reperfusion injury. Such information may be also useful in determining the favorable effect of treprostinil on remote organ damage. Although treprostinil administration holds great promise for attenuating myocardial injury in the course of hepatic ischemia-reperfusion injury, further research is warranted.
FOOTNOTES
Author contributions:Mouratidou C, Pavlidis ET, Katsanos G, and Tsoulfas G designed and performed the research; Mouratidou C, Pavlidis ET, Katsanos G, Kotoulas SC, Mouloudi E, Tsoulfas G, and Galanis IN analyzed data; Kotoulas SC, Mouloudi E, and Galanis IN contributed new analytic tools; Kotoulas SC, Mouloudi E, Tsoulfas G, Galanis IN, and Pavlidis TE reviewed the paper; Pavlidis TE approved the paper.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Greece
ORCID number:Christina Mouratidou 0009-0007-8657-2032; Efstathios T Pavlidis 0000-0002-7282-8101; Georgios Katsanos 0000-0002-5845-8175; Serafeim-Chrysovalantis Kotoulas 0000-0003-6092-1341; Eleni Mouloudi 0000-0003-0079-2012; Georgios Tsoulfas 0000-0001-5043-7962; Ⅰoannis N Galanis 0009-0001-4283-0788; Theodoros E Pavlidis 0000-0002-8141-1412.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wu RR
 World Journal of Gastrointestinal Surgery2023年9期
World Journal of Gastrointestinal Surgery2023年9期
- World Journal of Gastrointestinal Surgery的其它文章
- Preoperative and postoperative complications as risk factors for delayed gastric emptying following pancreaticoduodenectomy: A single-center retrospective study
- Comparative detection of syndecan-2 methylation in preoperative and postoperative stool DNA in patients with colorectal cancer
- Preoperative prediction of microvascular invasion in hepatocellular carcinoma using ultrasound features including elasticity
- Surgical management of gallstone ileus after one anastomosis gastric bypass: A case report
- Advances and challenges of gastrostomy insertion in children
- Surgical decompression for the management of abdominal compartment syndrome with severe acute pancreatitis: A narrative review