
MiR-873 regulates cell autophagy by targeting Beclin1 to promot inflammation and apoptosis of bronchial epithelial cells
2023-12-06 07:53CHENJieLINLingsangLISiguangMORubingDINGYipeng
Journal of Hainan Medical College 2023年15期
CHEN Jie, LIN Ling-sang, LI Si-guang, MO Ru-bing, DING Yi-peng,3✉
1.Hainan Hospital Affiliated to Hainan Medical University, Haikou 570311, China
2.Department of General Practice, Hainan General Hospital, Haikou 570311, China
3.Department of Respiratory and Critical Care Medicine, Hainan General Hospital, Haikou 570311, China
Keywords:
ABSTRACT Objective: Investigating the effects of miR-873 on apoptosis and autophagy in bronchial epithelial cells, as well as its regulatory role on Beclin1.Methods: Following transfection of miR-873 mimic into 16HBE cells for 48 hours, the mRNA level of miR-873 was quantified by qRT-PCR, and cell viability was evaluated by CCK-8 assay.The levels of IL-2, IL-6,IL-10, and TNF-α in the cell supernatant were determined using ELISA assay, while cell apoptosis was detected by TUNEL staining.LC-3 protein expression was examined by immunofluorescence, and mRNA and protein expression levels of Beclin1 were analyzed by qRT-PCR and Western blot, respectively.Moreover, dual-luciferase reporter gene technology was employed to investigate the binding site between miR-873 and Beclin1.Results:Transfection of miR-873 mimic into 16HBE cells significantly upregulated the mRNA level of miR-873, which led to the inhibition of cell proliferation and the promotion of secretion of pro-inflammatory cytokines IL-2, IL-6, and TNF-α, while suppressing the secretion of antiinflammatory cytokine IL-10.Moreover, miR-873 induced cell apoptosis and inhibited the expression of LC-3.Dual-luciferase reporter gene assay further confirmed the presence of binding sites between miR-873 and Beclin1 gene.Besides, miR-873 could target and suppress the mRNA and protein expression levels of Beclin1.Conclusion: miR-873 might modulate cell autophagy by targeting the Beclin1 gene, which can potentially promote inflammation and apoptosis in bronchial epithelial cells.
1.Introduction
Chronic obstructive pulmonary disease (COPD) is a common,preventable and treatable disease worldwide that is characterized by persistent respiratory symptoms and restricted airflow.The etiology of the disease is complex and is mainly caused by longterm exposure to harmful particles or gases, resulting in abnormal respiratory tract and/or alveoli[1].A large number of studies have shown that oxidative stress, inflammation, apoptosis, autophagy and aging are involved in the pathogenesis of COPD, in which inflammation and cell death are the most important pathogenic factors of COPD[2,3].Chronic persistent inflammatory response is considered to be the mechanism of airway structural injury,airway remodeling and small airway obstruction[4,5].Therefore,interventions targeting inflammatory responses and cell death may become effective strategies for COPD treatment.
MicroRNAs (miRNAs) are a class of small non-coding Rnas with a length of 18-22 nucleotides, which bind to the 3’ terminal non-coding region or coding region of the target mRNA and are involved in post-transcriptional inhibition, protein translation block and increased mRNA degradation in gene expression regulation[6].miRNAs are involved in a variety of biological processes, including regulation of cell proliferation, differentiation, apoptosis, autophagy,etc.[7].Numerous studies have shown that miRNAs are involved in regulating the transcription of multiple genes involved in lung function and inflammation[8,9].Based on the new perspective that miR-873 regulates autophagy by targeting Beclin1 gene to promote inflammation and apoptosis of bronchial epithelial cells, this paper will explain the correlation between miRNA and autophagy in the pathogenesis and development of COPD, providing theoretical basis for further understanding of the pathogenesis and development of COPD, and providing new ideas for clinical intervention in the progression of COPD.
2.Materials and methods
2.1 Reagents
Human bronchial epithelial cell 16HBE strain was purchased from Shanghai Cell Bank, Chinese Academy of Sciences.Fetal bovine serum, MEM medium, penicillin/streptomycin and pancreatic enzyme reagents from Invitrogen; Human interleukin-2 (IL-2),interleukin-6 (IL-6), human interleukin-10 (IL-10) and human tumor necrosis factor α (TNF-α) ELISA kits were purchased from Shanghai Renjie Biology.BCA protein quantitative detection kit, CCK-8 kit and TUNEL staining kit were purchased from Biyuntian Biological Company.Reverse transcription kit (P312)and real-time fluorescent quantitative PCR (Q331) were purchased from Novizan Biotechnology, Nanjing.Beclin1 and LC-3 primary antibodies were purchased from Abcam.The second antibody was purchased from Wuhan Bude Biological Engineering Co., LTD.; The double luciferase reporter gene was purchased from GenePharma in Shanghai.
2.2 16HBE Cell culture
16HBE cells were cultured in MEM medium containing 10% fetal bovine serum at a constant 37 ℃ and 5%CO2gas.The cells are passed down every two to three days.
2.3 miR-873 mimic transfected 16HBE cells
16HBE cells were inoculated into 6-well plates at a density of 2× 105cells/well and incubated at 37 ℃ and 5%CO2for 24 hours.miR-873 mimic (150 ng) was diluted with 100 µL serum-free MEM to produce a solution with a final concentration of 5 nmol.It was then mixed with 12 µL HiPerFect transfection reagent and left at room temperature for 10 min to form the transfection complex.The resulting transfection solution was added to each 2 mL medium hole.The cells were cultured at 37 ℃ for 72 h before follow-up experiments.
2.4 qRT-PCR detection
Integrated RNA was extracted from transfected 16HBE cells using Trizol reagent, and the concentration and purity of extracted RNA were evaluated by NanoDrop 2 000 (USA Thermo Feld).Reverse transcription kit was used to convert RNA into cDNA,and qPCR reaction was performed on ABI 7500QPCR instrument.The reaction conditions were as follows: predenaturation at 95℃for 5 min, denaturation at 95 ℃ for 5 sec, renaturation at 62 ℃ for 20 sec, extension at 72 ℃ for 30 sec, and 40 cycles.Using U6 and GAPDH as internal parameters, the expression levels of miR-873 and Beclin1 were calculated by 2-ΔΔct.miR-873 upstream primer:5′-GCAGGAACUUGUGAGUCUCCU-3′, miR-873 downstream primer: 5′-AGGAGACUCACAAGUUCCUGC-3′; U6 upstream primer: 5′-GCTTCGGCAGCACATATACTAA-3′; U6 downstream primer: 5′-AACGCTTCACGAATTTGCGT-3′; GAPDH upstream primer: 5′-TGTGTCCGTCGTGGATCTGA-3′; downstream primer:5′-CCTGCTTCACCACCTTCTTGA-3′;Beclin1 upstream primer:5′-CAGTACCAGCGGAGTAGTGA-3′;Beclin1 downstream primer:5′-TGTGGAAGGTGGCATTGAAGA-3′.
2.5 Detection of 16HBE proliferation using the CCK-8 method
The transfected 16HBE cells and control group cells were seeded in a 96-well plate at a density of 1 000 cells/well, and incubated at 37℃ with 5% CO2for 48 h.Then, 10 µL of CCK-8 reagent was added to each well (note the setting of control wells and blank background wells), and incubated in the dark at 37 ℃ for 2 h.The absorbance values of each well were measured at 450 nm wavelength using a spectrophotometer.When calculating cell proliferation rate, the following formula was used: Cell proliferation rate = (experimental group absorbance value - background value)/ (control group absorbance value - background value) × 100%.
2.6 ELISA kit detection
The transfected 16HBE cells and control group cells were seeded in a 6-well plate at a density of 2 × 105cells/well.After incubation at 37 ℃ and 5% CO2for 48 hours, the cell culture supernatant was collected using protein lysis buffer.Remove the kit 30 min in advance and balance to room temperature.During the experiment,100 µL of standard solution was added to the standard wells, 100µLof distilled water was added to the blank control wells, and 100µL of cell culture supernatant was added to the remaining wells.The OD values of each well were read at a wavelength of 450 nm according to the instructions of the ELISA assay kit.
2.7 TUNEL staining to detect apoptosis
The slide soaked in 75% alcohol and dried was placed in a 6-well plate, and the transfected 16HBE cells and control cells were inoculated at a density of 2 × 105cells per well.It was incubated at 37 ℃ and 5%CO2for 48 h, then rinsed with PBS and fixed with 4%PFA for 20 minutes.A solution containing 0.1% Triton X-100/0.1%sodium citric acid was added and incubated for 10 minutes,followed by protease K for 10 min.Droplets of TdT reaction mixture including TdT enzyme, dUTP-biotin marker and buffer were added to the sample and incubated for 1 hour at 37 ℃.Finally,Streptavidin-fluorescein was added to the sample and incubated at room temperature for 15 min without light.After washing with PBS,the sample was observed using a fluorescence microscope and the results were recorded.
2.8 Immunofluorescence detection of LC-3 expression
The cell treatment method referred to TUNEL staining, in which 0.1% Triton X-100 was used for perforation, and the cells were subsequently blocked with a 3% BSA solution at room temperature for 1 h.After treatment, the samples were washed with PBS and incubated with LC-3 primary antibody at 4 ℃ overnight.The following day, the samples were washed four times with PBST solution for 5 min each time.Then, fluorescently labeled secondary antibodies were added at room temperature for 2 h.Finally, DAPI was used for staining in the dark at 37 ℃ for 15 min, followed by another wash and mounting of the slides.The samples were observed and the results were recorded using a fluorescence microscope.
2.9 Western blot detection of Beclin1 expression
The transfected 16HBE cells and control group cells were seeded in a 6-well plate at a density of 3 × 105cells/well.After incubation at 37 ℃ and 5% CO2for 48 hours, the cells were washed with PBS once and lysed with 150 µL of lysis buffer containing 1% protease and phosphatase inhibitors per well.The protein concentration was detected using the BCA method.Then, the proteins were denatured with 5X SDS-PAGE loading buffer and heated in a 100 ℃ water bath for 10 min.The proteins were then separated by SDS-PAGE and transferred to a nitrocellulose membrane (PVDF).The membrane was blocked with 5% skim milk at room temperature for 2 hand incubated with Beclin1 primary antibody at 4 ℃ overnight.The primary antibody was recovered the next day and washed six times with PBST for 8 min each time.The membrane was then incubated with secondary antibody at room temperature for 1 h.After washing with PBST, the signal was detected using ECL staining technology.The Quantity One 4.52 image analysis software was used for image analysis, and the relative expression levels of the protein were calculated using GAPDH as an internal reference.
2.10 Dual-Luciferase Reporter Assay
In this study, a dual-luciferase reporter assay was performed in 293T cells by inserting the wild-type 3’ untranslated region (UTR)of Beclin1 and its mutant into the pMIR-RB-REPORT™ luciferase mRNA expression reporter vector.Next, 293T cells were seeded in a 6-well plate at a density of 5×104cells/well and transfected with Lipofectamine 2 000 with four groups: Beclin1-WT and miRNC, Beclin1-WT and miR-873, Beclin1-Mutant and miR-NC,and Beclin1-Mutant and miR-873.After 48 hours of transfection,the Firefly and Renilla luciferase activities were evaluated using a fluorescence spectrophotometer and normalized to the Renilla luciferase activity.
2.11 Statistical analysis
When using the statistical software GraphPad Prism 9.0 for data analysis, for metric data that conforms to a normal distribution,the sample mean ± standard deviation (±s ) is used to represent the data.The t-test is used for comparison of two groups, and oneway ANOVA is used for comparison of multiple groups.If the data conform to a normal distribution and have equal variances,the least significant difference (LSD) method is used for pairwise comparisons; if the variances are unequal, Dunnett’s T3 method is used for multiple comparisons.P<0.05 indicates a statistically significant difference.
3.Results
3.1 miR-873 inhibits bronchial epithelial cell proliferation and promotes its inflammatory response
Transfection of miR-873 mimic into 16HBE cells for 24 hshowed a significant increase in the mRNA level of miR-873 compared to the control group, as detected by qRT-PCR (3.44 ± 0.10vs1.03 ± 0.09,P< 0.001,t= 24.42), as shown in Figure 1A, indicating successful transfection of miR-873 mimic into 16HBE cells.
Transfection of miR-873 mimic into 16HBE cells for 24 h was followed by CCK-8 assay to measure cell viability.The results of the CCK-8 assay showed that, compared with the control group,the proliferation activity of 16HBE cells in the miR-873 mimic group significantly decreased from the second day (Day 2: 0.68± 0.06 vs1.16 ± 0.170, P < 0.001, t = 6.39; Day 3: 1.25 ± 0.12 vs 1.84 ± 0.05,P< 0.001,t= 7.86; Day 4: 1.81 ± 0.13vs2.5 ± 0.16,P< 0.001,t= 9.10; Day 5: 2.41 ± 0.20vs3.40 ± 0.12,P< 0.001,t = 13.11), as shown in Figure 1B, indicating that miR-873 mimic inhibits bronchial epithelial cell proliferation activity.
Transfection of miR-873 mimic into 16HBE cells for 24 hours was followed by ELISA assay to measure the expression of inflammatory factors IL-2, IL-6, IL-10, and TNF-α in the cell culture supernatant.The results showed that, compared with the control group, the expression levels of IL-2 (101.84 ± 3.15 vs 15.36± 1.422,P< 0.001,t= 30.65), IL-6 (64.01 ± 3.43vs11.19 ± 2,21,P< 0.001,t= 18.72), and TNF-α (163.93 ± 8.01vs76.37 ± 0.56,P<0.001, t = 31.04) were significantly increased, while the expression level of IL-10 (3.3 ± 0.21vs19.72 ± 1.26,P< 0.001,t= 5.82) was significantly decreased in the cell culture supernatant of the miR-873 mimic group, as shown in Figure 1C, indicating that miR-873 mimic induces inflammation response in bronchial epithelial cells.

Fig 1 Upregulation of miR-873 promotes inflammation in bronchial epithelial cells
3.2 miR-873 promotes apoptosis of bronchial epithelial cells
The TUNEL staining results showed that, compared with the control group, transfection of miR-873 mimic into 16HBE cells for 24 h significantly increased the number of apoptotic cells in 16HBE cells, as shown in Figure 2, indicating that miR-873 mimic promotes apoptosis of bronchial epithelial cells.
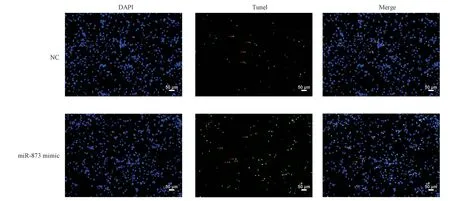
Fig 2 Upregulation of miR-873 promotes apoptosis in bronchial epithelial cells
3.3 miR-873 promotes autophagy of bronchial epithelial cells
The results of immunofluorescence staining showed that, compared with the control group, transfection of miR-873 mimic into 16HBE cells for 24 h significantly reduced LC-3 staining in 16HBE cells,as shown in Figure 3, indicating that miR-873 mimic inhibits autophagy of bronchial epithelial cells.
3.4 miR-873 targets and binds to Beclin1, inhibiting its expression
Transfection of miR-873 mimic into 16HBE cells for 24 h showed a significant decrease in the mRNA level of Beclin1 compared to the control group, as detected by qRT-PCR (0.41 ± 0.08 vs 1.02 ± 0.13,P< 0.001,t= 21.67), as shown in Figure 4A.Western blot analysis showed that, compared with the control group, transfection of miR-873 mimic into 16HBE cells for 24 h significantly inhibited the protein expression of Beclin1 (0.34 ± 0.04 vs 0.78 ± 0.09, P < 0.001,t= 14.48), as shown in Figure 4B.These results indicate that miR-873 can target and regulate the mRNA and protein expression of Beclin1.
Prediction from Targetscan database showed the existence of complementary binding sites between miR-873 and the 3’UTR of Beclin1, as shown in Figure 4C.Fluorescent luciferase reporter vectors containing the binding sites for Beclin1 3’UTR were cotransfected with miR-873 mimic or miR-873 inhibitor into 16HBE cells, and luciferase activity was measured after 24 h of incubation.The results showed that co-transfection of Beclin1 3’-UTR-WT and miR-873 significantly inhibited luciferase activity (0.35 ± 0.06 vs 1.03 ± 0.26,P< 0.001,t= 11.17), while co-transfection of Beclin1 3’-UTR-MUT and miR-873 had no significant effect on luciferase activity, as shown in Figure 4D, indicating the existence of a binding site between the 3’UTR of Beclin1 and miR-873.These results suggest that miR-873 inhibits the mRNA and protein expression of Beclin1 by targeting its binding site.
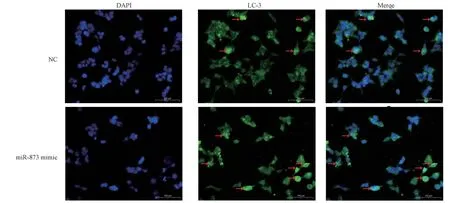
Fig 3 Upregulation of miR-873 suppresses autophagy in bronchial epithelial cells
4.Discussion
COPD is a common respiratory disease with a complex pathogenesis[2].In recent years, increasing evidence has shown that microRNAs (miRNAs) also play an important role in the progression of COPD.Studies have confirmed that miR-21 can induce activation of macrophages, neutrophils, and lymphocytes by downregulating the SATB1/S100A9/NF-κB signaling pathway,activating inflammatory response, and exacerbating the progression of COPD[10].Elevated expression of miR-223 in the lungs of COPD patients and smoke-induced mice promotes the progression of COPD through immune cell infiltration[11].Meanwhile, miR-125a-5p inhibits cigarette smoke-induced COPD/emphysema epithelial cell senescence by suppressing the Sp1/SIRT1/HIF-1a pathway,thus accelerating the progression of COPD[12].miR-150 has a protective effect on smoke-induced lung inflammation and can alleviate cigarette extract-induced apoptosis of airway epithelial cells [8].As a hypoxia-regulated miRNA, miR-125b participates in the acute exacerbation of COPD by regulating apoptosis of respiratory epithelial cells and inducing lung tissue damage[9].miR-126 promotes the release of inflammatory cytokines, leading to acute and chronic respiratory tract inflammation, which in turn causes pathological and physiological changes such as reversible airway limitation, airway remodeling, and airway hyperresponsiveness,thereby exacerbating the severity of COPD in patients[13].In this study, we found that transfection of miR-873 mimic into bronchial epithelial cells 16HBE significantly inhibited their proliferation activity, promoted apoptosis, induced secretion of inflammatory cytokines IL-2, IL-6, and TNF-α, and inhibited the secretion of IL-10.These results suggest that miR-873 participates in the regulation of COPD progression by regulating apoptosis and inflammatory response of bronchial epithelial cells.
Autophagy is a conserved lysosomal degradation pathway characterized by dynamic degradation and reprocessing of cell components to maintain physiological homeostasis[14].Autophagy as a kind of cell protection mechanism, can protect a variety of diseases, especially tumor, cardiovascular disease, neurodegenerative diseases and infectious diseases.However, over-activated autophagy can disrupt cellular homeostasis and lead to cell death[15].In addition, studies have shown that autophagy also plays a role in the pathogenesis of respiratory diseases, such as COPD, asthma and pulmonary failure[16].As one of the important factors regulating basic biological processes, miRNA is also involved in the regulation of autophagy.For example, miR-214-3p regulates ox-LDL-induced autophagy in HUVECs cells by directly targeting the 3’UTR of ATG5, thus participating in the regulation of atherosclerosis[17].Up-regulation of miR-93 enhances the killing effect of IR and TMZ on glioblastoma by inhibiting autophagy[18].In addition,upregulating miR-29c-3p inhibits autophagy and enhances the killing effect of cisplatin on ovarian cancer by targeting FOXP1/ATG14 [19].In this study, we found that upregulation of miR-873 can significantly reduce the expression of LC-3 in bronchial epithelial cells, suggesting that miR-873 also has the function of inhibiting autophagy.
miRNA is a type of small molecule RNA that affects the expression of target genes by binding to mRNA, and is involved in regulating multiple physiological processes such as cell life activities and development[20].miRNA forms RISC complex by combining with Argonaute protein, and binds with 3 ‘Untranslated region (3’ UTR) of mRNA in cytoplasm.On the one hand, miRNA RISC complexes can promote mRNA degradation, leading to a decrease in the expression of target genes; On the other hand, miRNA RISC complexes can also inhibit the translation process of mRNA, reducing protein synthesis of target genes[21].Beclin1 gene (BECN1) not only participates in the formation of Autophagosome, but also plays an important role in tumor genesis and development by regulating autophagic activity[22].The Bcl-2 family proteins Bcl-2, Bcl-xL, and Mcl-1 are anti apoptotic proteins that can bind to the Bcl-2 homologous 3 (borane)domain of Beclin 1 and play a rheostatic role in autophagy.Under normal circumstances, Beclin 1 binds to Bcl-2.When separated from Bcl-2, Beclin 1 can form PI3KC3 complexes with Vps15, Vps34(also known as type III PI3K), and other proteins such as Ambra 1 to regulate the initiation of autophagy[23].Studies have shown that miR-837-5p can inhibit the growth, invasion and angiogenesis of lung cancer cells by targeting the level of Beclin1, thus playing a role in inhibiting lung cancer metastasis[24].In this study, we used Targetscan database prediction and double luciferase reporter genes to verify the binding sites of miR-873 and Beclin1, and confirmed that miR-873 mimic could target and inhibit the mRNA and protein expression of Beclin1.Therefore, we concluded that miR-873 may inhibit the autophagy of bronchial epithelial cells by targeting Beclin1 gene, promote inflammation and apoptosis of bronchial epithelial cells, and participate in the occurrence and development of COPD.
Authors’ contribution
CHEN Jie: Double luciferase experiment and writing; LIN Lingsang: Cell culture and transfer; LI Si-guang: Cell proliferation,apoptosis and autophagy detection; MO Ru-bing: RT-PCR and western blot detection; DING Yi-peng: Review and financial support.All authors agree to contribute to this manuscript without conflict of interest
 Journal of Hainan Medical College2023年15期
Journal of Hainan Medical College2023年15期
- Journal of Hainan Medical College的其它文章
- Establishment of extensively drug-resistant Pseudomonas aeruginosa pneumonia model in rat
- Monitoring and analysis of contamination of Vibrio parahaemolyticus and Vibrio alginolyticus in seafood in Haikou
- Research progress on cardiotoxicity mechanism of doxorubicin and prevention and treatment of traditional Chinese medicine
- Study on regulating mechanisms of oxocrebanine obtained from Stephania hainanensis H.S.Lo et Y.Tsoong on microtubule sites and tubulin in human breast cancer MCF-7 cells
- Effect of acupuncture on acupoint "Yingxiang-Hegu" on Th1, Th2 cytokines and T-bet/GATA-3 of allergic rhinitis rats
- Study on the mechanism of Fuzi in the treatment of allergic rhinitis based on network pharmacology and experimental validation