
SOD1基因p.H44R罕见位点突变致肌萎缩侧索硬化症1例报告并文献复习
2023-12-18 04:55王雅欢刘洪雨何金婷王姣琦
中风与神经疾病杂志 2023年11期
王雅欢, 杨 偲, 刘洪雨, 何金婷, 王姣琦
运动神经元病(motor neuron disease,MND)是一组起病隐匿的慢性进行性神经系统变性疾病,病因尚不十分清楚,可能与遗传或环境因素相关。该病主要侵犯大脑皮质锥体细胞、脑干运动神经核及脊髓前角运动神经元。肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)是最常见的一种,根据是否有家族史分为家族性肌萎缩硬化症(familial amyotrophic lateral sclerosis,fALS)和散发性肌萎缩硬化症(sporadic amyotrophic lateral sclerosis,sALS)。该病的早期表现是进行性肌萎缩、肌无力和延髓麻痹,最终常累及呼吸肌导致呼吸衰竭死亡。5%~10%的患者有家族史,但大多数患者的病因仍不清楚[1]。该病不可逆转,且缺乏有效治疗手段,其诊断为排除性诊断,需与表现相似的其他运动神经元病相鉴别[2],早期诊断困难。现将吉林大学中日联谊医院神经内科收治的1 例以右下肢疼痛、无力为首发症状,通过肌电图及全外显子测序诊断的SOD1罕见位点突变致ALS 病例进行回顾分析及文献复习,希望对该病在临床上的早期诊断及预后评估有所帮助。
1 病例资料
患者,男,40 岁,因右下肢疼痛5 个月,无力4 个月,于2022年2月入院,5 个月前接种疫苗后出现右下肢肌肉酸痛,因未影响肢体活动而未在意,4 个月前出现右下肢无力、肉跳,逐渐发展并出现臀部、大腿及小腿肌肉萎缩,服用B 族维生素类药物后症状未见缓解,近期使用筋膜枪理疗,下肢无力的症状仍逐渐加重,入院前20 d出现右腿上台阶费力,其余肢体无明显异常。病程中患者无头晕、无智能减退、无动作迟缓、无腰部疼痛、无舌肌萎缩、无吞咽及呼吸困难,体重未见明显变化。既往无基础疾病。家族史:患者堂哥及2 个姑姑被诊断为“运动神经元病”,但均已过世,无法提供临床资料及相关检查结果。患者父亲因肺癌于48岁去世(患者家系图见图1)。

图1 患者家系图
入院后神经系统查体:意识清楚,颅神经查体未见明显异常,右下肢屈髋肌力2级,伸髋肌力3级,屈膝肌力3 级,伸膝肌力4 级,远端背屈肌力2 级,跖屈肌力4 级,余肢体肌力未见异常,右下肢肌张力减弱,肌容积减少,右下肢膝腱反射减弱,跟腱反射未引出,无感觉异常,双侧病理反射阴性,余查体未见异常。
实验室检查:肌酸激酶600 U/L(正常值<164 U/L),肌酸激酶同工酶17.00 ng/ml(正常值2.00~7.20 ng/ml),肌红蛋白149 ng/ml(正常值23.00~112.00 ng/ml),D-二聚体3 340.00 ng/ml(正常值80~500 ng/ml)。甲功3 项、降钙素原、C 反应蛋白、常规免疫、肿瘤标志物、免疫球蛋白+补体、风湿免疫相关抗体未见明显异常。头部核磁未见异常。腰骶丛神经MR 平扫+增强:左侧骶1、2 神经根束膜囊肿可能性大,腰椎MR:腰5-骶1 椎间盘轻度突出。肺部CT:左肺下叶外基底段结节,性质倾向良性。腹部超声:脂肪肝。肌电图示:右下肢呈神经源性改变(根或以上水平受损可能);左胫前肌、左腓肠肌可见神经源性改变(见图2)。进一步完善的腰穿脑脊液检查示脑脊液无色透明,细胞总数4×106/L,白细胞数4×106/L,血抗神经节苷脂抗体阴性。提检全外显子测序,结果回报前初步诊断为下运动神经元综合征,待除外肌萎缩侧索硬化症。给予患者营养神经、针灸、理疗、被动康复训练等对症治疗,患者自觉右踇趾背屈稍有改善后出院。
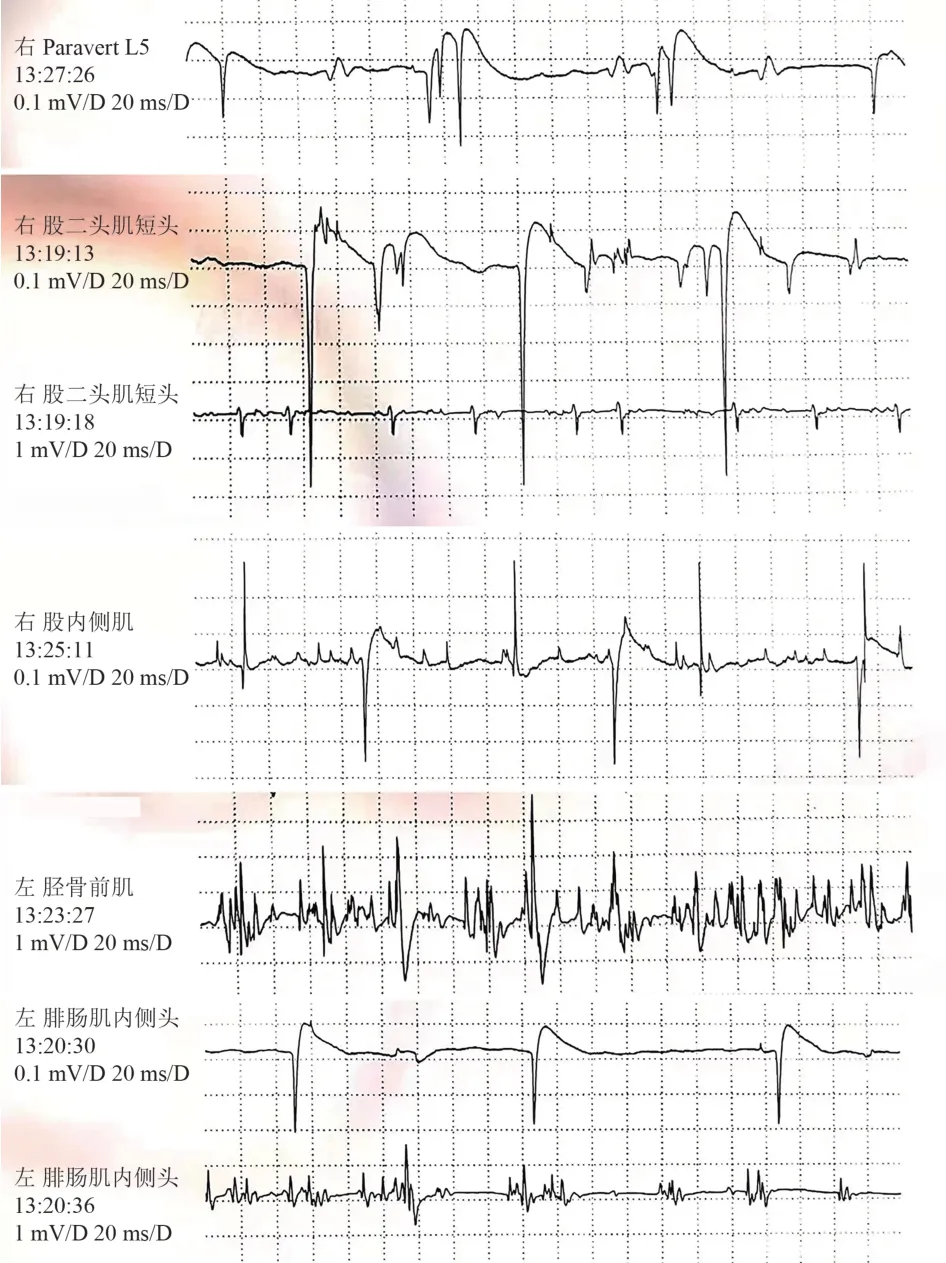
图2 患者的肌电图结果
全外显子测序提示患者携带SOD1基因上的一个杂合错义变异:c. 131A>G:p. H44R。由于先证者父母未能配合验证,变异来源未知。该变异为cDNA的第131 位碱基由A 替换为G,导致SOD1基因第44位密码子由编码组氨酸变为编码精氨酸(见图3)。
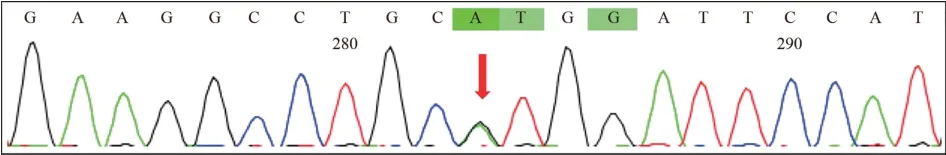
图3 患者的一代测序峰图
获得全外显子测序结果后,从下运动神经元综合征入手剖析患者的诊断。患者无中毒、辐射,无脊髓灰质炎、艾滋病、肠道及水痘带状疱疹病毒感染史,无蜱叮咬史,脑脊液细胞数正常,可除外中毒、感染、辐射等病因,重点筛查免疫、变性及遗传方面。患者偏侧受累,抗神经节苷脂抗体阴性,不符合吉兰-巴雷综合征(guillain-barré syndrome, GBS);患者肿瘤标志物阴性,腹部超声、胸部CT 筛查未见占位性病变,暂不支持副肿瘤性下运动神经元综合征;患者肌电图无局灶性运动传导阻滞,且早期出现的明显的肌肉萎缩也不支持多灶性运动神经病(multifocal motor neuropathy,MMN),故可除外免疫因素。变性方面,单肢肌萎缩(monomelic muscular atrophy,MMA)为自限性,多见于青少年,累及上肢时又称平山病,该病很少累及对侧肢体,与患者的发病年龄及肌电图上左下肢亚临床神经源性损害不符。遗传方面,遗传性远端运动神经病(distal hereditary motor neuropathy,dHMN)及脊髓性肌萎缩(spinal muscular atrophy,SMA)为单基因病,全外显子测序结果不支持。最后,从遗传变性的角度需要鉴别ALS 和进行性肌萎缩(progressive muscular atrophy,PMA)。PMA主要表现为进行性肌无力或萎缩,不伴上运动神经元功能障碍,该病比其他类型运动神经元病的进展慢,暂不能完全除外。患者全外显子测序提示携带SOD1基因上的一个杂合错义变异。目前国内尚未将基因检测纳入ALS 诊断标准,但是查阅文献发现该突变是ALS 已知的致病突变,呈常染色体显性遗传。2015年修订的E1 Escorial 诊断标准中提出,若基因检测发现患者携带已知的肌萎缩侧索硬化症致病基因,且具有一个区域的上运动神经元或下运动神经元损害证据可诊断为ALS,如果至少有一个一级或二级亲属患有ALS 则考虑为遗传性ALS[3]。患者查体见下运动神经元损伤证据,并有基因检测支持,故排除PMA,诊断为ALS。嘱患者服用利鲁唑,随访。后患者就诊于北京大学第三医院,维持ALS诊断。2 个月后电话随访,患者出现了左下肢的无力。
2 讨 论
ALS 常中年以后隐蔽起病,进展缓慢,是一种上、下运动神经元同时受累的神经系统变性疾病,通常以肌无力、萎缩等为主要表现,受累部位常有肌束震颤,伴有腱反射亢进、病理反射阳性。大多数患者最终因呼吸衰竭死亡。目前国际公认的ALS诊断标准共有3 种,依次为E1 Escorial 诊断标准、Airlie House 诊断标准和Awaji-shima 电生理诊断标准,目前临床上应用最广的是Airlie House 诊断标准,该标准将ALS 诊断等级分为确诊级肌萎缩侧索硬化、拟诊级肌萎缩侧索硬化、实验室支持拟诊级肌萎缩侧索硬化、可能级肌萎缩侧索硬化[4]。基于以上诊断标准,极大地提高了ALS 患者的诊断效率和病情评判。根据患者的临床表现,最新的ALS 分型将肌萎缩侧索硬化分为8型[5]:(1)经典型,以上肢或下肢肌无力为首发症状,锥体束征常不明显,是男性患者中最常见的类型,平均发病年龄在62.8 岁,10年生存率为13%,伴发额颞叶痴呆(frontotemporaldementia,FTD)的占4%;(2)延髓型,以构音障碍和(或)吞咽困难等延髓损害为特点,发病6 个月内锥体束征可不明显,随着疾病发展,锥体束征可越来越明显,男性发病率略低于女性,10年生存率仅为3.4%,FTD发生率最高为9%;(3)连枷臂型,以上肢近端肌无力和萎缩起病,可伴有上肢的腱反射活跃及Hoffman征,肌张力正常,发病后,功能受累必须局限于连枷肢体至少12个月,男女发病比例为4∶1,10年存活率为17.4%,很少伴发FTD,仅占1.4%;(4)连枷腿型,主要为双下肢进行性肌无力和萎缩,病程中下肢可有腱反射活跃或Babinski 征,肌张力正常。注意除外无远端受累的患者,这种情况常提示为经典型。男女发病比例为1.03∶1,10年存活率为12.8%,合并FTD 的占4.1%;(5)锥体束型,以锥体束征为主,如严重的痉挛性截瘫或四肢瘫,可出现在疾病早期或晚期,常累及至少两个不同区域,出现明显的下运动神经元损害的体征,如肌肉无力和萎缩。肌电图检查存在慢性和活动性的失神经损害。此种表型发病较早,常于58.3 岁左右,男女比例为1.04∶1,FTD较少见,占2.5%,10年存活率为31.9%;(6)呼吸型,此类型患者普遍存在呼吸障碍,表现为呼吸困难或端坐呼吸,发病半年内脊髓或球部受累症状较轻,可伴有上运动神经元损害迹象。该型为最罕见的表型,男性发病率0.06/10 万,女性发病率0.01/10 万,无1例患者存活10年以上;(7)纯下运动神经元综合征(pure lower motor neuron syndrome,PLMN),病变累及脑神经运动核和脊髓前角细胞,典型表现为肌无力或萎缩、腱反射减弱,但感觉不受累,男性发病率是女性2 倍,不伴有FTD,预后与其他表型相比较好,10年生存率为36.6%;(8)纯上运动神经元综合征(pure upper motor neuron syndrome,PUMN),临床表现包括痉挛性截瘫和(或)四肢瘫,病理反射亢进,腱反射活跃,言语障碍等,此种表型发病率较低(男女均为0.12/10 万),存活时间最长(13.1年),10年生存率为71.1%。此外还有学者认为原发侧索硬化(primary lateral sclerosis,PLS)和进行性肌萎缩(progressive muscular atrophy,PMA)为ALS 的特殊类型,PLS 较为少见,一般中年起病,4年内仅有上运动神经元受累表现;PMA仅有下运动神经元受累表现,以男性患者多见,发病年龄晚,平均生存期比ALS显著延长。以上两种类型随着疾病的发展会出现上、下运动神经元同时受累的情况,与ALS表现相似,所以上述两种类型可以被归为ALS 的特殊类型[4]。ALS复杂的临床分型不仅说明了该病在起病形式、受累模式、流行病学等方面的多样性,而且也提示了该病病因的复杂性。
目前发现与fALS发病相关的基因约30多种,包括铜/锌超氧化物歧化酶(SOD1)基因、反式激活反应-DNA 结合蛋白(TARDBP)基因、肉瘤熔合(FUS)基因等[6]。其中,SOD1基因突变是我国sALS 和fALS 最常见的突变类型,在sALS 患者中占1%~2%,fALS 占25%[7]。1993年Rosen 等首次发现SOD1突变引发ALS[8]。SOD1是一种由153 个氨基酸组成的酶,在神经系统、肝脏及红细胞中高表达,参与自由基清除。目前已知的与ALS 相关的SOD1基因突变超过150个[9]。本文列举了常见的几种SOD1基因突变类型、遗传方式及临床表现(见表1)。不同突变型引发的ALS在起病时间、受累部位、病程进展速度等方面都存在差异。其中A4V突变是最常见的SOD1基因突变,常表现为下运动神经元损伤,平均存活时间仅1.5年。D90A突变提示上运动神经元损伤较下运动神经元损伤明显,且疾病进展较慢,患者存活时间相对较长。G37R、G41D、G93C突变提示患者存活期较长,G37R、L38V、L106V突变提示患者发病年龄较小。其中,L106V基因突变患者的发病年龄是目前已知最年轻的,为35.5 岁,而I113T基因突变的发病年龄最晚,为58.9 岁[10]。大多数ALS 患者在症状出现后3~5年内死亡,但变异性很大,有些患者在发病后几个月死亡,而有些患者甚至存活了20年。即使是来自于一个家族的不同个体,虽然具有完全相同的基因突变位点,但其生存期和发病年龄上也有很大的差异,这说明还存在其他改变表型的因素有待进一步发现[9]。

表1 常见SOD1基因突变位点及其遗传方式、临床表现
本例患者以右下肢疼痛、无力伴肌肉萎缩起病,感觉系统无阳性体征,症状暂未累及其他肢体,且无球部损伤症状,定位在脊髓前角及前根。肌电图示右下肢神经源性改变(根或以上水平),左侧胫前肌、腓肠肌可见自发电。该肌电图结果一方面支持上述定位;另一方面也提示左下肢出现亚临床的急性神经源性损害。患者腰骶丛核磁、腰椎核磁未见明确的脊髓及神经根病变,故除外神经根损伤,定位在脊髓前角细胞。患者病前有疫苗接种史,有可疑ALS家族史,定性上应重点考虑免疫因素及遗传变性病。根据下运动神经元综合征的病因分析,患者免疫相关等实验室检查结果未见异常,影像学及脑脊液检查未见炎性改变,暂不支持免疫炎症,故遗传变性病的可能性大,不除外以下运动神经元损伤起病的ALS。患者存在ALS 家族史,虽然无法明确证实,但从一元论的角度考虑,基因测序有望为患者早期诊断提供依据。当患者出现下运动神经元综合征的表现并疑诊ALS 时,完善非受累肢体或节段的肌电图检查可提供临床前的下运动神经元损伤证据,为了解疾病全貌,对疾病进行早期诊断提供帮助。全外显子测序提示患者SOD1基因突变,最终诊断为ALS。该患者为杂合子突变,文献报道ALS突变类型大多数是常染色体显性遗传,D90A、D96N基因突变是常染色体隐性遗传或常染色体显性遗传[23],基因检测结果提示患者为SOD1基因第二外显子c. 131A>G:p.H44R突变,该变异为罕见变异。关于H44R基因突变致ALS的报道最早可以追溯到1995年[24]。2003年首次对该突变致ALS 的临床表现进行了详细的描述,该患者是名58 岁的日本女性,因下肢单瘫就诊,有ALS家族史,发病7个月后死于呼吸衰竭。该患者家族系谱显示为常染色体显性遗传,具有完全外显率[25]。2017年,该基因突变位点在我国患者中首次报道,1 例47 岁男性以上肢无力起病,9 个月后因呼吸衰竭死亡,不同的是此患者无家族史,该变异为SOD1基因上的一个杂合错义突变[7]。本例患者的发病年龄较前2 例更早,单肢瘫起病,临床症状虽然局限,但肌电图已提示其他肢体的早期受累。目前患者病程较短,还有待进一步随访以明确患者病情进展及预后情况。结合已报道的病例特点,推测H44R基因突变导致的ALS 可能主要以下运动神经元损伤为主,可有球部受累症状,病情进展快,生存期可能较短。
尽管目前美国FDA已批准利鲁唑作为ALS的首选治疗药物,但其疗效一般,仅延长2~3 个月的无气管切开术生存期[26],患者最终走向死亡。2022年我国ALS诊断和治疗专家共识提示基因检测阳性可加速ALS 的诊断进程,使患者尽早开始接受药物治疗[27]。随着测序技术的发展,基因检测的经济成本越来越低,该技术对广大民众的可及性也在提高。在使用该技术早期诊断治疗的同时,我们也要警惕技术滥用及随之而来的伦理问题。
伦理学声明:本研究经由吉林大学中日联谊医院伦理委员会审批(审批号:2023111001)。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:王雅欢负责采集数据、数据整理及分析、论文撰写;杨偲、刘洪雨、何金婷负责采集数据、分析数据、研究指导;王姣琦负责研究指导、论文修改。
猜你喜欢
考试与评价·高二版(2020年2期)2020-09-10
现代电生理学杂志(2016年3期)2016-07-10
现代电生理学杂志(2016年4期)2016-07-10
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中外医疗(2015年16期)2016-01-04
实用手外科杂志(2015年1期)2015-08-27
现代电生理学杂志(2015年3期)2015-07-18
现代电生理学杂志(2015年2期)2015-07-18
西安交通大学学报(医学版)(2015年2期)2015-02-28
中医研究(2013年9期)2013-03-11
