
利用WGCNA挖掘黄精属植物多糖合成通路关键基因*
2023-12-21 04:52陈雪梅
吉首大学学报(自然科学版) 2023年5期
汪 蓉,孟 然,陈雪梅,孟 盈
(吉首大学生物资源与环境科学学院,湖南 吉首 416000)
黄精属(PolygonatumMill.)植物是多年生草本植物,隶属于天门冬科洛林亚科黄精族[1],该属在我国广泛分布,可药食两用,常见于南北方不同的中医药派系及相关食谱中[2-4].在最新版《中国药典》中,中药黄精具体是指黄精(PolygonatumsibiricumDelar. ex Redoute)、多花黄精(PolygonatumcyrtonemaHua)和滇黄精(PolygonatumkingianumCollett &Hemsl.)3个种[4].其中黄精主要分布在我国北方,滇黄精主要分布于我国西南地区,多花黄精广泛分布在国内多个地区且已有人工培育居群[5-6].现代药理学及市场应用中,黄精及相关提取物主要用于抗肿瘤、抗氧化、抗衰老等领域,并有较好的疗效.黄精的主要活性成分有多糖、皂苷、黄酮等化学成分,其中糖类较多,以黄精多糖和低聚糖为主[7-10].另外,黄精属中的玉竹(Polygonatumodoratum(Mill.) Druce)也是重要的中药资源.最新研究表明,玉竹的主要活性成分具有降血糖、抗炎、调节免疫和抗肿瘤作用[11].黄精属植物具有重要的医药价值,当前研究方向主要集中在其药理活性、提取工艺和人工培育技术等方面[12-14].在后基因组时代,黄精属植物仍然缺乏高质量的全基因组数据作为参考.目前,在有效成分的调控机制方面,部分研究人员基于转录组数据及相关算法,发现了黄精属植物中多糖有效成分的多个调控基因,这为黄精属植物种质资源的保护与合理开发利用奠定了研究基础[14-16].随着测序技术的提升,这个方向的研究也在不停地推进与深化.
生物信息分析技术的更新让转录组学的分析方案不再局限于差异的查找与定位,越来越多的研究强调通过整合多个因素解决更加具体的科学问题[17-19],以便在基因数据中找到一些高度关联的基因表达网络来解释复杂的生物学现象.近年来,加权基因共表达网络分析(Weighted Gene Co-expression Network Analysis,WGCNA)被广泛应用于多个研究领域,其可通过无监督的机器学习内核,将不同样本的表达量数据进行表达网络计算,并根据基因表达量的相关性划分模块,分析这些相对独立的模块与分组之间的关系来定位相关靶基因.本研究基于黄精属中多花黄精和玉竹的根状茎及叶片的转录组数据,采用加权基因共表达网络分析方法,结合KEGG数据库中多糖合成相关通路信息,计算核心基因在多花黄精、玉竹的叶片及根状茎之间表达模式的相关性,以获取影响黄精属植物多糖类有效成分合成的关键基因.
1 材料和方法
1.1 数据获取与分析
本研究以黄精属下多花黄精和玉竹2个物种根状茎和叶片的转录组数据开展后续分析.在NCBI中筛选SRP166090和SRP170979队列的相关样本,共计14个.其中SRP166090是多花黄精的转录组测序结果,基于Illumina HiSeq 4000平台完成建库测序,选取其中的根状茎(SRR8074960)和叶片(SRR8074961)样品参与分析[20];SRP170979是玉竹的转录组测序结果,同样基于Illumina HiSeq 4000平台完成转录组建库测序,筛减其中的非目标器官数据,共保留12个样品[21].使用Aspera软件下载所有的fastq文件,并通过FastQC软件对原始数据进行质量评估,然后使用Fastp软件过滤掉低质量的reads和接头序列.
使用Trinity软件完成转录组组装,然后执行CD-HIT-EST脚本过滤组装结果中的冗余转录本.基于liliopsida_odb10数据库,使用BUSCO软件完成组装结果的质量检测,并通过BLAST2GO软件对组装结果进行注释.用align_and_estimate_abundance.pl软件调用RSEM分析不同样品的表达定量,同时用FPKM值来衡量不同测序深度造成的表达量差异.
1.2 构建加权基因共表达网络
使用WGCNA 1.69(R 4.1)软件包进行加权基因共表达网络构建,以均一化后的样本基因表达矩阵及样品来源组分信息作为输入数据,利用表达量最高的8 000个基因构建共表达网络.为进行WGCNA计算中的软阈值筛选,通过pickSoftThreshold函数来比较不同软阈值参数下相关性的差异,选择“Power=12”计算最终的无尺差异邻接矩阵.基于该矩阵数据进一步计算拓扑有差异的矩阵,利用动态剪切算法将基因进行聚类分析.通过比较不同模块与分组信息之间的相关性,挑选出最为相关的模块,利用Module Membership及Gene Significance参数过滤模块中的基因.
1.3 多糖合成通路相关性
通过KEGG数据库的注释数据,筛选WGCNA分析结果中的核心基因,提取其中与多糖合成通路相关的基因.使用R中的limma软件包对所有基因进行筛选,挑选分组间存在显著表达差异的基因.差异基因的挑选标准设定为P_value<0.05,并且logFC阈值服从整体数据分布的95%分位数.将差异基因的表达量矩阵按照分组信息拆分,使用R中的cor ()函数计算表达矩阵的相关性,执行ggpubr软件包中的ggscatter函数来查验这些基因在根状茎数据与叶片数据间的皮尔森相关性系数.
2 结果
2.1 组装与注释
黄精属植物作为传统中药,具有重要的药用价值,但是由于其基因组庞大,全基因组测序数据组装困难,目前仍然缺乏高质量的全基因组数据.在本研究中,笔者将所有样品的原始数据合并后使用Trinity软件进行无参组装.组装得到的结果共计207 Mb,其中共获得了394 149条转录本,GC占比43.25%,N50值664 bp.BUSCO分析共匹配到数据库中3 236个基因,其中单拷贝基因1 853个(占比57.26%),多拷贝基因969个(占比29.94%),还有277个仅部分比对上的基因,以及137个未比对到数据库的基因.笔者在多个数据库中进行比对注释,大量转录本被比对到多糖合成相关通路的关键基因序列上,如糖酵解/糖异生(ko00010)、淀粉和蔗糖代谢(ko00500)、氨基糖和核苷酸糖代谢(ko00520)等通路.表1列出了多糖合成的相关通路及相应的基因数量.此外,本研究还进行了NR,eggNOG,KEGG,GO数据库的注释,注释结果如图1所示.

表1 多糖合成通路

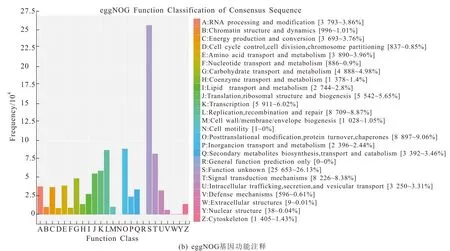


图1 非冗余组装结果注释信息Fig. 1 Annotation Information of Unigenes
2.2 加权基因共表达网络构建
笔者在表达量矩阵中选择高表达的前8 000基因进行分析,有效避免低表达基因对分析过程的影响.软阈值计算结果分布如图2所示.由图2可知,当Power值为12时,R2数值大于0.85,且平均连接度小于100,分布较为合理,因此选择该软阈值进行后续分析.

图2 软阈值计算结果分布Fig. 2 Soft Threshold Calculation Result Distribution
WGCNA结果如图3所示.在动态剪切算法分析中,共获得9个基因模块.其中:Turquoise模块包含的基因数量最多,共3 386个;其次为Blue模块,包含1 689个基因;Pink模块最少,仅包含54个基因;Grey模块功能为回收未能分配的基因,包含880个基因;其余模块中所包含的基因数量为100~900个.在9个基因模块中,选择同时满足|r|>0.5,P<0.05的Turquoise模块作为本研究的相关特异性模块,并对模块中的基因进行过滤筛选,要求基因与分组信息的关联性同时满足Membership值大于0.8且Gene Significance值大于0.4,最后得到1 608个核心基因.此外,本研究也选取了Blue模块(|r|=0.55,P=0.04),但与Turquoise模块相比,Blue模块中基因与分组信息的关联性较低,因此过滤标准设定为Membership值大于0.6且Gene Significance值大于0.4,根据实际数据共筛选出核心基因1 753个.


图3 WGCNA结果Fig. 3 Result of WGCNA

图4 叶片与根状茎之间的差异基因Fig. 4 Differential Expression Genes Between Leaves and Rhizomes
2.3 差异基因及相关性分析
为了进一步验证多糖合成通路相关基因在根状茎与叶片中表达模式的相关性,笔者挑选出在叶片和根状茎间表达模式高度相关的基因.叶片与根状茎之间的差异基因如图4所示.由图4可知,本研究在3 236个基因中共找到88个上调基因和14个下调基因.笔者将这102个基因的序列放在NCBI数据库中检索,确定了基因名称和基因对应的功能.表2列出了多糖合成通路的显著关联基因.从表2可以看出,多糖相关合成通路中的2个关键基因是果糖二磷酸醛缩酶(Fructose-Bisphosphate Aldolase,FBA)和肌醇半乳糖苷合成酶(Galactinol Synthase,GOLS),它们在根状茎与叶片之间显著差异表达.
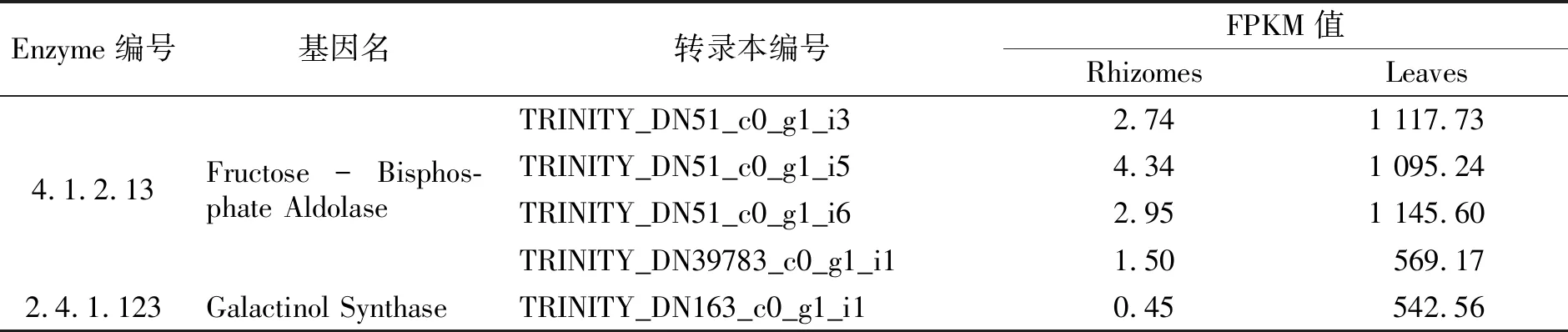
表2 多糖合成通路显著关联基因
多糖合成基因在根状茎与叶片中的相关性分析结果如图5所示.根据基因表达矩阵,笔者计算了样品间的皮尔森相关性系数.皮尔森相关性热图见图5(a).为了更好地呈现2组数据间关键基因表达模式的相关性,笔者计算了每个基因在不同分组中的相关性系数平均值,并绘制线性回归图(图5(b)).在95%的置信区间内,这些基因在根状茎和叶片之间的表达量具有很高的相关性(R=0.88,P=8.5×10-5).

图5 多糖合成基因在根状茎与叶片中的相关性分析Fig. 5 Correlation Analysis of Polysaccharide Synthesis Genes Between Rhizomes and Leaves
3 讨论
黄精属植物是多年生草本植物,入药仅使用地下的根状茎部分,其有效成分的检验需要挖掘根状茎进一步提取分析,因而无法进行广泛有效检测[22-24].当前已经广泛使用的转录组测序方案中提出的基因表达量与表型之间的调控关系,为解决该困境提供了行之有效的方法.本研究通过获取黄精属下多花黄精和玉竹2个物种多个样本的转录组数据,利用WGCNA方法,在Blue和Turquoise模块中筛选出5 075个与分组相关的基因,另外还查找到与FBA基因和GOLS基因相对应的多个转录本,并根据这些转录本在根状茎与叶片的表达模式之间的皮尔森系数,确定了2个基因的表达模式在器官间具有显著关联性(R=0.88,P=8.5×10-5),为后续基于叶片转录组数据预测根状茎中多糖成分含量提供了研究基础.
FBA基因在多个物种中均有深入研究,并且经过大量的实验验证,确认该基因及相关蛋白是糖酵解、糖异生、磷酸戊糖通路中的重要原件,其在光合作用中也是卡尔文循环的重要参与者[25].FBA基因常常被富集到糖酵解/糖异生(ko00010)、戊糖磷酸途径(ko00030)和半乳糖代谢(ko00052)等与多糖合成直接相关的通路中,这对于判断黄精中多糖相关有效成分含量具有指示性作用.此外,该基因也被报道在棉花、玉米等物种不同器官中参与了多糖类化合物的合成调控[26-27].本研究结果显示,半乳糖代谢通路(ko00052)中的GOLS基因差异表达,并与叶片和根状茎之间的表达模式高度相关.目前针对GOLS基因的研究主要围绕该蛋白处于非生物因素下对植物生长的影响方面,在不同的影响因素下,GOLS基因的应答机制存在明显的异质性[28-29].这说明植株生境受到影响时,GOLS基因能够反映植株的生长状态,为进一步确定有效成分含量提供判断依据.综上,FBA基因和GOLS基因在根状茎与叶片间的表达模式为黄精属植物多糖有效成分的观测提供了分子指示物.
随着多组学时代的到来,植物研究领域中数据量大爆炸的时代也已到来,如何更好地应用大数据来解决科学问题,是当前最大的科研需求.本研究采用加权共表达网络分析方法整合了多个转录组数据,从全新角度为黄精属植物有效成分的观测提供了分子证据.尽管在当前分析中仍然存在缺乏黄精属植物的高质量基因组数据,以及部分现有数据无法使用的问题,但转录组数据分析方案的多样性可基本弥补这些缺陷.虽然FBA基因参与多糖类化合物的合成调控在多个物种中均有验证,但是在黄精属植物中的实际调控过程不明,后续仍需要通过分子生物学实验来进一步验证.
猜你喜欢
生物技术通报(2023年10期)2023-11-23
草业学报(2022年8期)2022-11-23
Digital Chinese Medicine(2020年3期)2020-11-03
云南农业科技(2020年5期)2020-10-14
中国现代医生(2019年5期)2019-04-25
天然产物研究与开发(2018年6期)2018-07-09
中成药(2018年3期)2018-05-07
药学与临床研究(2015年4期)2015-06-05
中成药(2014年11期)2014-02-28
中国粮油学报(2014年7期)2014-02-06
