
Three previously undescribed metabolites from Cordyceps cicadae JXCH-1,an entomopathogenic fungus
2023-12-29 13:42JingFanPaiLiuKuanZhaoandHePingChen
Jing Fan,Pai Liu,Kuan Zhao and He-Ping Chen
Abstract Three previously undescribed compounds,cordycicadione (1),cordycicadin F (2),and 7-hydroxybassiatin (3),were isolated from the cultures of Cordyceps cicadae JXCH1,an entomopathogenic fungus.Their structures and relative configurations were elucidated primarily by NMR spectroscopic analysis.The absolute configurations of 1 and 2 were determined by ECD calculations.Single-crystal X-ray diffraction method was adopted to determine the absolute configuration of 3.Compound 2 is a polycyclic polyketide with an unusual enol ether moiety and a spiro ring.The compounds obtained in this study were subjected to screening their inhibition against the proliferation of the human lung cancer cell line A549 and the production of nitric oxide in murine macrophages RAW264.7.
Keywords Insect pathogenic fungus,Cordyceps cicadae,Steroid,Polyketide,Cyclic dipeptide
1 Introduction
The genusCordyceps,a group of insect-pathogenic fungi,includes many species with important medical applications in Chinese Medicine.The sensu stricto species used in Chinese Medicine areCordyceps sinensis,Cordyceps militaris,andCordyceps cicadae[1].Among the three species,C.sinensishas been used for treating diverse chronic diseases for more than 300 years,whileC.militarisis a species which has been widely used as a materia medica,but also used a food supplement.C.cicadaeis a parasitic fungus formed by cicada nymphs infected by the fungusC.cicadae(=Isaria cicadae) [2].Jin Chan Hua,a fungus-insect complex used in Chinese Medicine,is one of the earliest medicines recorded in classics of Traditional Chinese Medicine.Pharmacological experiments results suggested thatJin Chan Huais effective remedies for convulsions,chronic kidney diseases,measles,asthma,insomnia,and heart palpitations [3].However,although the medicinal values ofC.cicadaehave long been recognized,but unlike theC.militaris,theC.cicadaeare not cultivatable [4],and thereby,its shortage of supply has hampered the widely application of this species.
Recently,there have been few studies on the chemical composition and pharmacological effects of theC.cicadaestrains,which have facilitated the alternative application of the crude fractionated products of this fungus.It is clear that the bioactive substances ofC.cicadaeinclude nucleosides,polysaccharides,sterols,cyclic dipeptides,amino acids,and other small organic compounds [5-8],which have extensive pharmacological effects including immunomodulatory,antitumor [9],neuroprotective [10],antibacterial [11],and anti-diabetic [12] activities.However,in terms of the exact chemical profiles of the natural products derived from this fungus,only a handful of compounds were reported.Cordycicadins A-D,purified from the fermentation broth of the fungusC.cicadaeJXCH1,are unprecedented C20polyketides with unusual exocyclic enol ether bridges [3].Biological evaluations of these polyketides indicate that they showed significant antifeedant activity against silkworm larvae,implying the potential of these compounds in green agrochemicals development.Therefore,the secondary metabolites ofC.cicadaeare indeed worth to be further investigated.Herein,we report the purification as well as structural elucidation of three metabolites from the cultures of the fungusC.cicadaeJXCH1.
2 Results and discussion
Cordycicadione (1,Fig.1) was obtained as a pale-yellow oil.It presented an ion peak with highest abundance atm/z497.28726 [M+Na]+in the (+)-HRESIMS analysis,which returned a molecular formula of C28H42O6(calcd for C28H42O6Na,497.28736),corresponding to eight degrees of unsaturation.The structural elucidation of1was accomplished by thorough analysis of the NMR spectra.The1H NMR spectroscopic data of1recorded in methanol-d4(Table 1) showed resonances for two CH3singlet protons atδH1.16 (Me-18),1.28 (Me-19),and four CH3doublets atδH0.74 (d,J=6.9 Hz,Me-28),0.76(d,J=6.9 Hz,Me-26),0.93 (d,J=7.0 Hz,Me-27),1.08 (d,J=6.9 Hz,Me-21),four oxygen-attached methine protons atδH3.30 (overlapped,H-23),3.76 (s,H-12),4.19(br.d,J=7.6 Hz,H-22),4.49 (m,H-16).The13C NMR as well as DEPT spectroscopic data of1recorded in methanol-d4(Table 2) displayed sharp carbon signals that were classified into six CH3,six CH2,nine CH [four oxygenated CH atδC74.8 (C-23),80.3 (C-12),83.5 (C-16) and 85.2 (C-22)],two double bond carbons,three quaternary carbons [sp3hybridized,one oxygenated,δC83.7 (C-14)],and two ketone carbons atδC201.0 (C-11) and 214.1(C-3).The 1D NMR spectroscopic data of1displayed similarity with the data of asperfloroid [13],an ergostane steroid purified from the fungusAspergillus flocculosus16D-1 which resides in sponge,except for the substitutions at C-3,C-5,C-6 and C-7.The C-5 and C-6 are double carbons of the reference compound asperfloroid,while C-5 of1is a methine,C-6 and C-7 are methylenes.These changes were substantiated by1H-1H COSY spectrum that showed correlations of H2-4/H-5/H2-6/H2-7(Fig.2).The HMBC correlations from H2-1,H2-2,and H2-4 to C-3 atδC214.1 suggested that C-3 was a ketone carbon in1,instead of being a hydroxymethine in asperfloroid (Fig.2).Analysis of the ROESY spectrum recorded in dimethyl sulfoxide-d6allowed to determine the relative configuration,which enabled the presence of some pivotal exchangeable protons.The ROESY correlations of H-12/Me-18/OH-14 indicated that OH-14 was β orientation,and OH-12 was α orientation.Moreover,the diagnostic ROESY correlations of Me-18/H-20,Me-18/H-15β(δH1.82),Me-18/H-22,and H-15α (δH2.20)/H-16/H-17 suggested theS*andR* configurations of C-16 and C-22,respectively (Fig.2).The relative stereochemistry of both C-23 and C-24 were assigned asR* by comparing the1H and13C NMR chemicals shifts with those of asperfloroid.Based on above-mentioned analysis,the ECD calculation method was adopted to assign the absolute configuration of1.As a result,the calculated ECD of 5S,10S,12R,13S,14S,16S,17R,20S,22R,23R,24Rshowed compatible curve with that of the experimental CD spectrum (Fig.3).Therefore,the evidence presents in above texts help to establish the structure of compound1(Fig.1).

Fig. 1 The chemical structures of compounds 1-3

Table 1 1H NMR Spectroscopic data of compounds 1-3 (600 MHz)
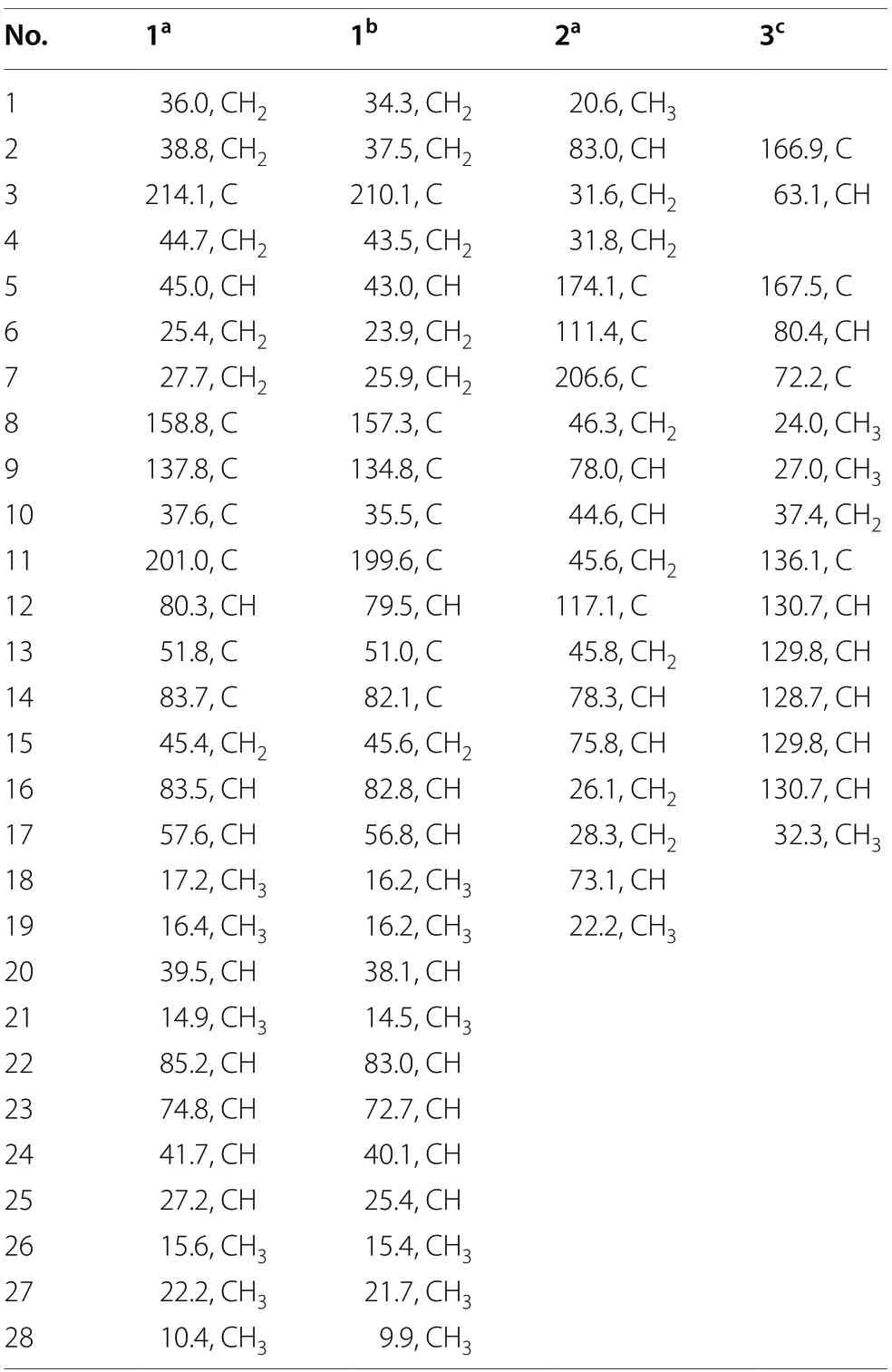
Table 2 13C NMR spectroscopic data of compounds 1-3(150 MHz)
Cordycicadin F (2,Fig.1),a pale-yellow oil,possesses the molecular formula of C19H26O5from the ion peak atm/z335.18518 [M+H]+(calcd for C19H27O5,335.18530)in (+)-HRESIMS analysis.The1H NMR spectroscopic data of2(Table 1) showed two doublet methyl signals atδH1.14 (d,J=6.2 Hz,Me-19),1.40 (d,J=6.2 Hz,Me-1),five oxygen-connected methine protons atδH3.41 (m,H-18),3.89 (m,H-15),3.99 (dd,J=4.9,2.1 Hz,H-14),4.62 (overlapped,H-9),4.68 (m,H-2).The13C NMR along with the DEPT spectroscopic data of2(Table 2)indicated resonances for 19 carbons attributable to two CH3carbon atδC20.6 (C-1) and 22.2 (C-19),seven CH2carbons atδC26.1 (C-16),28.3 (C-17),31.6 (C-3),31.8(C-4),45.6 (C-11),45.8 (C-13),46.3 (C-8),six methines[five oxygenated ones atδC73.1 (C-18),75.8 (C-15),78.0(C-9),78.3 (C-14),and 83.0 (C-2)],one pair of double bonds atδC111.4 (C-6) and 174.1 (C-5),one ketal carbon and one ketone carbon atδC117.1 (C-12) andδC206.6 (C-7),respectively.These data of2(Tables 1 and 2)resembled those of the known compound opaliferin [14],a polyketide isolated from the entomopathogenic fungusCordycepssp.NBRC 106954.The difference between compound2and opaliferin is that the C-14 and C-18 of opaliferin are each substituted with a hydroxy group,while the C-14 and C-18 of2are attached by an ether bond.This change was reinforced by the HMBC correlations from H-18 to C-14 (Fig.2).The tetrahydrofuran moiety (C-1-C-5) was established by HMBC correlations from H-2,H2-3,and H2-4 to C-5.The presence of cyclopentanone moiety (C-6-C-10) was confirmed by the HMBC correlations of H-8,H-9 to C-6,C-7,and of H-10,H-11 to C-6.The HMBC correlation from H2-4 to C-6 and chemical shifts of C-5 (δC174.1),C-6 (δC111.4)in13C NMR spectrum suggested the existence of an enol moiety.The HMBC correlations from H2-11 to C-12,H2-13 to C-11,H2-13 to C-12,H-14 to C-12,H-15 and H-9 to C-12,together with the chemical shift of C-12 (δC117.1) indicated that C-12 was an acetal carbon which connected to C-9 and C-15 by two epoxy bonds.All of these data were highly similar to opaliferin.Furthermore,the configurations of the chiral carbons of2were consistent with those of opaliferin,which were assigned as 2R,5E,9S,10R,12R,14S,15S,18Rby biosynthetic considerations,comparison with the NMR data,and the ROESY correlations of H-14/H-15/H-18,and H-9/H-10 (Fig.2),and confirmed by ECD calculation (Fig.4).Therefore,compound2was established and was trivially named cordycicadin F.
The molecular formula of the colorless crystals 7-hydroxybassiatin (3,Fig.1) was determined as C15H19NO4by the protonated ion peak atm/z278.13851[M+H]+(calcd for C15H20NO4,278.13868) in the HRESIMS analysis,requiring seven degrees of double bond equivalence.The proton NMR data of3(Table 1)showed three singlet methyls [two methyl protons atδH1.02 (Me-9) and 1.07 (Me-8),oneN-methyl protons atδH3.05 (Me-17)],two methine protons atδH3.10 (s,H-6) and 4.67 (t,J=4.8 Hz,H-3),one pair of methylene protons atδH3.28 (dd,J=14.2,4.8 Hz,H-10),3.37 (dd,J=14.2,4.8 Hz,H-10),one exchangeable proton atδH4.80 (s,OH-7),and five aromatic protons.The13C NMR and DEPT spectroscopic data of2(Table 2) presented carbon signals for three methyl carbons atδC24.0 (C-8),27.0 (C-9) and 32.3 (C-17),one methylene carbon atδC37.4 (C-10),seven methines [one oxygenated atδC80.4(C-6),the others atδC63.1 (C-3),128.7 (C-14),129.8(C-13),129.8 (C-15),130.7 (C-12),130.7 (C-16)],two quaternary carbons atδC72.2 (C-7) and 136.1 (C-11),and two ketone carbons atδC166.9 (C-2) and 167.5(C-5).The aforementioned data exhibited high similarity to bassiatin,which was isolated from the cultured broth of the fungusBeauνeria bassianaK-717 [15].The 2D NMR spectroscopic analysis revealed that the major structural discrepancy between3and bassiatin was that the substituted pattern of C-7.The C-7 of bassiatin is a methine while C-7 of3is attached by a hydroxy group.The difference was fully corroborated by the HMBC correlations from OH-7 to C-6,C-7 and C-9.Therefore,the absolute configuration of3was assigned as 3S,6Rby single-crystal X-ray diffraction analysis with the Flack parameter of 0.01(4) (Fig.5).Hence,the structure of compound3was established (Fig.1),and trivially named 7-hydroxybassiatin.

Fig. 2 Key 2D NMR correlations of compounds 1-3
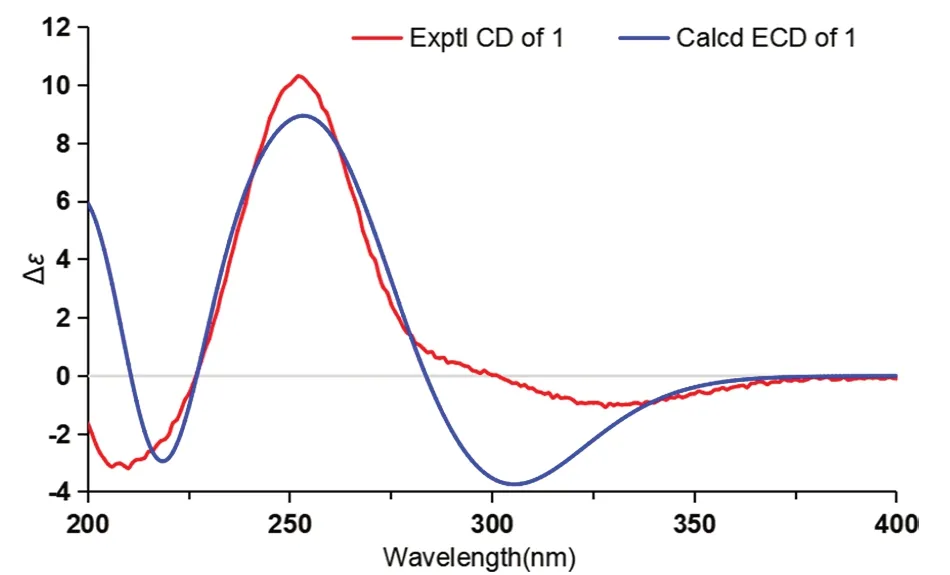
Fig. 3 Experimental CD and calculated ECD of 1

Fig. 4 Experimental CD and calculated ECD of 2
The compounds isolated were tested for their cytotoxicity toward A549 cell line (human lung cancer),and the anti-inflammatory activity in murine RAW264.7 macrophages by evaluating the production of nitric oxide(NO).However,the results suggested that they were devoid of any activity.
3 Conclusion
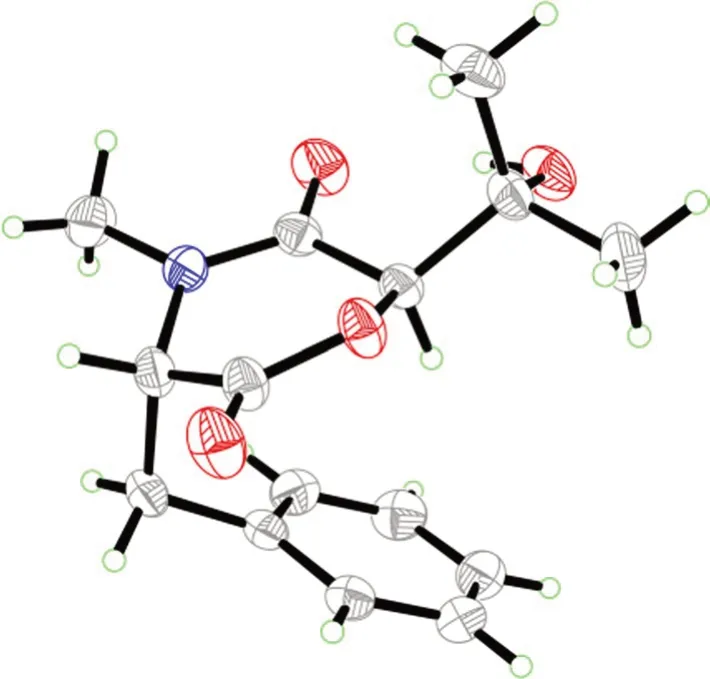
Fig. 5 X-ray ORTEP drawing of compound 3
Three new compounds isolated from the cultures of the entomopathogenic fungusC.cicadaeJXCH1.The isolates in this study were subjected to screening their inhibition against the proliferation of the A549,a human lung cancer cell line,and the production of nitric oxide in murine macrophages RAW264.7.Although they were devoid of any activity in the biological activity screening model used in this study,but these compounds may play important or untapped roles during the process of fungal invasion into the insect hosts.Besides,this study extends the diversity of secondary metabolites in the fungusC.cicadae,which also promotes the understanding of chemical basis of traditional Chinese medicine.In subsequent experiments,we also hope to find molecules with novel skeleton or better activity fromC.cicadaeto provide more lead compounds for drug research and development.
4 Experimental section
4.1 General experimental procedures
The laboratory instruments used in this study are as same as those reported in the literature [16-18].
4.2 Fungal material
A flower-like cicadae larva-fungi complex was collected in Dajueshan Natural Reserve,Fuzhou,Jiangxi Province,China,in June 2018.The “flower” part of the complex was wash by disinfectant distilled water for three times.The inner part of the “flower” which contained powder-like spores was transferred onto the potatodextrose-agar (PDA) plate supplemented with ampicillin and streptomycin.The fungus was purified by several times of efforts,and it was identified by sequencing the internal transcribed spacer region,which was amplified from the genomic DNA of this fungus with the primers ITS1 and ITS4.As a result,the sequencing data (Gen-Bank Accession No.OP591391) suggest that the fungus wasC.cicadae.A voucher specimen of the cicadae larvafungi complex and the fungus (JXCH1) was kept by the Mushroom Bioactive Natural Products Research Group of South-Central Minzu University.
The fresh strain ofC.cicadaeJXCH1 was grew on PDA medium for seven days (25 ℃).Then the plate agar was cut into small cubes to inoculate 200 Erlenmeyer flasks (500 mL),each containing 250 mL of culture medium which consists of 5 g glucose,1.5 g pork peptone,5 g yeast extract,0.5 g KH2PO4,0.5 g MgSO4per liter.The flasks were cultured on dark shakers at 25 °C and 160 rpm for 25 days.
4.3 Extraction and isolation
Based on previous experience on handling the cultures of this fungus [3],50 L of methanol was added into the cultures and soaked for more than 3 days in room temperature,followed by centrifugation to break apart the mycelia and broth.The liquid phase was evaporated in vacuo to 3 L,and it was further partitioned with ethyl acetate (EtOAc) (3 × 3 L).The EtOAc layer was gathered and concentrated to give the extract A (20.2 g).The mycelia part was soaked in 5 L MeOH (3 × 3 days).The MeOH extract was combined and removed in reduced pressure,then dissolved in water and partitioned against EtOAc to give the extract B (33.3 g).The total crude extract (A+B,53.5 g) was fractionated by MPLC with a stepwise gradient of MeOH-H2O (20:80-100:0)to afford fifteen fractions (A-O).
Fraction I was subjected to a size-exclusion CC(Sephadex LH-20) eluting with acetone and yielded ten subfractions (I1-I10).Subfraction I2 further separated by prep-HPLC (H2O-MeCN: 75:25-50:50,25 min,4 mL min-1) to gain six subfractions (I2a-I2f).Subfraction I2b was separated into three subfractions (I2b1-I2b3)by prep-HPLC (H2O-MeOH: 60:40-40:60,30 min,3 mL min-1).Subfraction I2b1 was purified on prep-HPLC (H2O-MeCN: 69:31-57:43,15 min,4 mL min-1)to yield compounds3(tR=14.5 min,3.9 mg).
Fractions K-M were combined and subjected to a size-exclusion CC (Sephadex LH-20) eluting with acetone and yielded fifteen subfractions (K1-K15).Compounds1(tR=23.26 min,2.4 mg) and2(tR=19.55 min,3.0 mg) were isolated from subfraction K6 by prep-HPLC (H2O-MeCN: 70:30-46:54,30 min,4 mL min-1).
4.4 Characterization data
4.4.1 Cordycicadione (1)
Pale-yellow oil;[α]23.0D +46.9 (c0.05,MeOH);UV(MeOH) λmax(logε) 245.0 (3.50) nm;1H NMR(600 MHz,CD3OD and DMSO-d6) data,see Table 1,13C NMR (150 MHz,CD3OD and DMSO-d6) data,see Table 2;HRESIMSm/z497.28726 [M+Na]+(calcd for C28H42O6Na,497.28736).
4.4.2 Cordycicadin F (2)
4.4.3 7-Hydroxybassiatin (3)
4.5 Computational details
The Gaussian 16 package was used as the software for all the calculation jobs [19].Firstly,conformational analyses of the candidate molecules were searched at MMFF94s force field.The returned conformers which occupy more than 1% proportion were optimized by the density functional theory (DFT) method at the B3LYP/6-31G(d) level.The optimized conformers were subjected to electronic CD calculations.The method used in ECD calculation was time-dependent DFT (TD-DFT) at B3LYP/6-31G(d,p) level,the solvent model was IEFPCM model in MeOH (Additional file 1).The ECD calculation results were processed by the SpecDis software (v1.71)[20],the calculated ECD and experimental CD curves were plotted in the Microsoft Office Excel program.
4.6 Single-crystal X-ray diffraction data of 3
A colorless block-like sample with the molecular formula of C15H19NO4,M=277.31,approximate dimensions 0.228 mm × 0.247 mm × 0.322 mm,was used for the X-ray crystallographic analysis on the BRUKER D8 QUEST diffractometer.The integration of the data using an orthorhombic unit cell yielded a total of 20,646 reflections to a maximumθangle of 79.51° (0.78 Å resolution),of which 3218 were independent (average redundancy 6.416,completeness=99.4%,Rint=3.45%,Rsig=2.93%) and 3160 (98.20%) were greater than 2σ(F2).The final cell constants ofa=6.7753(4) Å,b=12.8266(8)Å,c=17.1279(11) Å,α=90.00°,β=90.00°,γ=90.00°,volume=1488.48(16) Å3,T=273(2) K.Data were corrected for absorption effects using the Multi-Scan method (SADABS).The structure was solved and refined using the Bruker SHELXTL Software Package,using the space groupP212121,with Z=4,μ(CuKα)=1.54178.The final anisotropic full-matrix least-squares refinement onF2with 189 variables converged at R1=2.87%,for the observed data andwR2=7.66% for all data.The goodness-of-fit was 1.053.The absolute configuration was determined by the Flack parameter=0.01(4),which was determined using 1303 quotients [(I+)-(I-)]/[(I+)+(I-)][21].The crystallographic data have been deposited to the Cambridge Crystallographic Data Centre (CCDC 2250285).
4.7 Biological activity assays
4.7.1 Anti-proliferative assay
The anti-proliferative of the isolated compounds in this study against the five cancer cell lines were conducted by the same protocol in Ref.[22].
4.7.2 Anti-inflammatory activity assay
The anti-inflammatory of the isolated compounds against the murine RAW264.7 macrophages were conducted by the same protocol in Ref.[22].
Supplementary Information
The online version contains supplementary material available at https:// doi.org/ 10.1007/ s13659-023-00410-2.
Additional file 1: Figure S1.1H NMR spectrum of compound1(600 MHz,CD3OD).Figure S2.13C and DEPT NMR spectra of compound1(150 MHz,CD3OD).Figure S3.HSQC spectrum of compound1(CD3OD).Figure S4.1H-1H COSY spectrum of compound1(CD3OD).Figure S5.HMBC spectrum of1(CD3OD).Figure S6.HRESIMS report of compound1.Figure S7.1H NMR spectrum of compound1(600 MHz,DMSO-d6).Figure S8.13C and DEPT NMR spectra of compound1(150 MHz,DMSO-d6).Figure S9.HSQC spectrum of compound1(DMSO-d6).Figure S10.1H-1H COSY spectrum of compound1(DMSO-d6).Figure S11.HMBC spectrum of compound1(DMSO-d6).Figure S12.ROESY spectrum of compound1(DMSO-d6).Figure S13.1H NMR spectrum of compound2(600 MHz,CD3OD).Figure S14.13C and DEPT NMR spectra of compound2(150 MHz,CD3OD).Figure S15.HSQC spectrum of compound2(CD3OD).Figure S16.1H-1H COSY spectrum of compound2(CD3OD).Figure S17.HMBC spectrum of compound2(CD3OD).Figure S18.ROESY spectrum of compound2(CD3OD).Figure S19.HRESIMS report of compound2.Figure S20.1H NMR spectrum of compound3(600 MHz,CD3COCD3).Figure S21.13C and DEPT NMR spectra of compound3(150 MHz,CD3COCD3).Figure S22.HSQC spectrum of compound3(CD3COCD3).Figure S23.1H-1H COSY spectrum of compound3(CD3COCD3).Figure S24.HMBC spectrum of compound3(CD3COCD3).Figure S25.ROESY spectrum of compound3(CD3COCD3).Figure S26.HRESIMS report of compound3.Figure S27.CD spectrum of1.Figure S28.CD spectrum of compound2.Figure S29.CD spectrum of compound3.ECD calculation details of compounds1and2.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant number 81903512) and the Fundamental Research Funds for the Central Universities,South-Central Minzu University (Grant Number CPT22033).The authors thank the Analytical & Measuring Center,School of Pharmaceutical Sciences,South-Central Minzu University for the spectra test.
Author contributions
KZ and HPC designed the experiment;JF and PL performed the isolation and identification of all the compounds;PL measured the NMR spectra;JF wrote the original manuscript;PL,KZ,and HPC revised the manuscript.
Availability of data and materials
All data generated and analyzed during this study are included in this published article and its Additional file 1.
Declarations
Competing interests
The authors declare that there is no competing interests or personal relationships that could affect the work reported in this article.
Received:29 August 2023
Accepted:11 October 2023

 Natural Products and Bioprospecting2023年6期
Natural Products and Bioprospecting2023年6期
- Natural Products and Bioprospecting的其它文章
- Quinones from Cordia species from 1972 to 2023: isolation,structural diversity and pharmacological activities
- Ginsenoside compound-K attenuates OVX-induced osteoporosis via the suppression of RANKL-induced osteoclastogenesis and oxidative stress
- The alkynyl-containing compounds from mushrooms and their biological activities
- A recent update on development,synthesis methods,properties and application of natural products derived carbon dots
- Natural product rhynchophylline prevents stress-induced hair graying by preserving melanocyte stem cells via the β2 adrenergic pathway suppression
- Kaemtakols A-D,highly oxidized pimarane diterpenoids with potent anti-inflammatory activity from Kaempferia takensis