
基于特征结构组合描述的抗癌药物筛选
2024-01-05 12:49杨亚鑫王璟德
华东理工大学学报(自然科学版) 2023年6期
杨亚鑫, 王璟德, 孙 巍
(北京化工大学化学工程学院, 北京 100029)
癌症逐渐成为了人类主要死亡原因,据柳叶刀统计,2017 年中国人十大死因中,癌症就占了四个席位[1]。癌症具有早期症状不明显、易扩散和转移、导致人体免疫力下降等危害。目前,以化学药物为代表的化疗是治疗癌症的一大重要手段,但是化疗药物具有副作用强、易产生抗药性、损伤免疫系统等不足之处,因此需要继续开发副作用小、选择性强,不易耐药,对免疫系统损伤更小的抗癌药物。
开发新抗癌药物有从现有化合物中筛选新抗癌药物和设计新抗癌药物分子两种方式。其中,筛选新抗癌药物是基础,而新抗癌药物的设计需要对具有药效的先导化合物(或药物)的结构和药效基团有所了解[2],相对复杂,因此现在大多采用从现有化合物中筛选这一方式来寻找新抗癌药物。从现有化合物中筛选新抗癌药物主要分为以实验筛选和计算机虚拟筛选两种为代表的高通量筛选技术。
高通量筛选技术是使用自动化设备同时快速测试数千到数百万样本的生物活性并筛选样本的实验过程。高通量筛选药物技术应用较广、较为成熟,但仍然依赖于微量的药物实验、高灵敏度检测系统,而且具有随机性,耗费大量时间和金钱[3]。相比于实验筛选方法,计算机虚拟筛选可以预先在海量的化合物库中筛选出潜在的抗癌活性物质,减少需要实验的药物种类,从而节约时间、人力和物力。
使用计算机虚拟筛选抗癌药物的前提有两个:一是必须将药物分子处理成计算机可以识别的格式,这一点借助分子表征算法可以做到;二是基于数字格式把握抗癌药物和非抗癌药物之间结构和性质的差异,因此又产生了表示分子结构和性质的分子描述符或分子指纹。由于表示时出发角度不同以及分子本身结构、性质多样性,分子指纹或描述符种类也具有多样性。鉴于结构决定性质的认知,选用表征结构的分子指纹或描述符为描述药物分子结构的基础。
目前使用的表征分子结构和性质的分子指纹或描述符较多,而不同分子指纹或者描述符对筛选抗癌药物的效果也不尽相同。研究者们采用的多种分子指纹[4]包括Pubchem 指纹(801 个)、分子存取系统指纹(MACCS,166 个);分子描述符包括Mordred 分子描 述 符(Mordred ,1 826 个) 、RDKit 分 子 描 述符(RDKit,139 个)等。目前常采用机器学习算法如随机森林、遗传算法、支持向量机递归特征消除、或相似性分析等特征选择方法对分子指纹或描述符进行重要性分析选择来减少个数[5-7]。大多数研究者对分子指纹或者描述符个数的精简和挑选并不重视,筛选标准过于宽松,往往挑选后得到的分子指纹或描述符数量仍然多达数十至数百个。付洺宇等[8]采用商 业 软 件Molecular Operating Environment和 Python开源库Mordred 分别计算了365 个和1 613个描述符,只去除了空值和非数值型描述符,筛选后计算得到的非空值、纯数值描述符仍分别多达278 个和882 个。杜雪平[9]则使用方差过滤和Lasso 回归消除数据集中的噪声特征和关联特征,但过滤后仍有50 个特征。Yang 等[10]直接采用Chemical Development Kit 平台计算了默认参数的药物哈希指纹1 024 个,并以此作为评价结构相似性基础,但未对其进行特征筛选;Li 等[11]则只对数量较多的Mordred 描述符进行了特征筛选,未对MACCS和Morgan2 指纹(1 024个)进行筛选。数量过多的分子指纹或描述符增加了构建模型的复杂度, 并增加了对抗癌药物活性结构的提取难度。
由于Pearson 相关系数能评估变量线性关系,卡方检验能检验分类变量之间的关联,因此本文结合了这两种相关特征选择方法,从数据相关性角度计算分子指纹或描述符之间的相关性,去除不独立描述符,按照分子指纹或描述符与药物类别的关系排名,并对它们进行较大限度的个数精简,选择筛选效果较好的方案,将原本数量级超过102的指纹或描述符精简至101数量级,以达成用尽量少的描述符个数从众多药物中有效筛选抗癌药物的目的。Pearson 相关系数和卡方检验这两种方法的结合,可以从已有的描述符或分子指纹中快速去除关联性较大的分子指纹或描述符,选择出对抗癌药物筛选贡献最大的分子指纹或描述符,表示出描述抗癌药物最重要的特征结构组合,从而更具有针对性地筛选出抗癌药物。本研究不仅加快了抗癌药物的筛选和开发、明确未知药物有潜力的研究方向,还有望探究抗癌药物中对药效贡献最大的特征,为针对性设计抗癌药物提供坚实的基础。
1 实验部分
1.1 数据库和软件工具
数据库:Drugbank (https://go.drugbank.com/classyfication)是一个包含超过10 000 种药物或潜在药物的化学、药理学、医学以及分子生物学信息的药物信息数据库。Pubchem (https://pubchem.ncbi.nlm.nih.gov)则是关于有机小分子的化学物理性质、生物活性数据库,并且有较为详细的文献支撑。
软件工具:Python(版本:3.8) 的RDKit(版本:2022.03.1)库是一个用于化学信息学的开源工具包,它可用于化合物描述符和指纹生成、化合物结构相似性计算等。
1.2 抗肿瘤药物结构和性质的数字表征
为了方便计算机运算处理种类多样、结构复杂的化合物分子,需要把化合物分子转化成计算机容易处理的各种数据格式,并对化合物分子的结构和性质进行数字化描述,为计算机虚拟筛选提供基础。
目前化合物分子数字化格式分为图、字符串、连接表、矩阵[12]等。简化分子线性输入系统(Simplified Molecular Input Line Entry System, SMILES)使用频率最高,它是使用常见的字母和符号来编码分子结构的线性字符串,例如,它使用大写化学元素符号表示脂肪链上的原子,“=”表示双键等。图表示方式使用图形表示分子,把原子或基团看作点、化学键看成边,可以直观地显示原子的排列和结合方式。矩阵表示方式是用矩阵形式表示分子组成和连接方式,常用的有原子邻接矩阵,能表示每个原子与其他原子的连接情况,它是稀疏矩阵,相比图更加方便处理。连接表则是建立其他数据表相互关系的表格,常用的连接表包括sdf 文件,mol2 文件等,其较为详细表示原子和化学键的属性以及它们的关系,还包含分子的部分性质。
由于图的计算机处理方法往往较为繁琐,常用的原子邻接矩阵中冗余较多,连接表涉及较多表格信息,表示关系过于抽象,而字符串研究较多,使用广泛,并且节约空间,容易计算和检索,故本文采用SMILES 为药物分子数据表达格式。通过这些分子数字化格式可以计算出各种分子描述符或分子指纹。
分子描述符是逻辑和数学程序运行后得到的最终结果,它能把分子数字表征编码的化学信息转化为有用数字或实验结果[5]。分子描述符主要表述分子的结构或者性质,包括分子组成、拓扑结构几何信息等。分子指纹是一串离散的数字串,主要检验某个特定子结构是否存在,且子结构定义与分子指纹种类有关。目前使用的描述符个数较多,并且对抗癌药物筛选影响不同,可能存在冗余描述符,又因为结构决定性质,因此,本文以MACCS 指纹、RDKit 描述符、Mordred 描述符为基础,从中筛选出表征分子结构的抗癌特征描述符组合。
MACCS 是开源的166 位[11]二进制字符串,预定义了一系列子结构,每个指纹位数代表着一个特定子结构,比如第42 位代表氟原子,第99 位代表碳碳双键。指纹位数若出现字符“0”代表特定化合物中该子结构不存在,“1”代表该子结构在特定化合物中存在。该分子指纹定义的子结构含义清晰明确,可解释性高,且方便运算。
RDKit 是RDKit 库中自带的139 个描述符,包含分子组成描述符、分子连接性描述符、拓扑结构描述符等,计算简便,对分子结构概括性强。
Mordred[13]是在RDKit 的基础上对描述符进一步扩充完善后得到的描述符集合,可计算1 826 个描述符,除了RDKit 所包含的种类外,还包含邻接矩阵描述符、自相关描述符、几何描述符等,种类多,数量多,涉及描述符范围较广泛、较为全面。
本文基于收集的药物样本集,对Drugbank 和Pubchem 数据库中的抗癌药物和非抗癌药物进行分类标记,并且采用上述分子指纹或描述符形成初始数据集,再利用相关特征筛选方法精简分子指纹或描述符,结合决策树算法进行分类,找出对筛选抗癌药物效果较好的分子指纹或描述符,运用相关化学知识对结果做出归纳,总结出筛选抗癌药物的分子指纹或描述符的条件。
1.3 实验步骤
1.3.1 数据获取 通过Drugbank、Pubchem 数据库获取相关药物,确定抗癌药物,将剩余药物处理为非抗癌药物,形成有标记的药物数据集。本文搜集了11286种药物,其中抗癌药物201 种,其余均认为是非抗癌药物。将抗癌药物标记为“1”,非抗癌药物标记为“0”。获取了有标记的药物数据集后,通过Python 中的RDKit 安装包获取每个药物分子的标准SMILES,用于MACCS、RDKit、Mordred 的计算,计算出的分子指纹或描述符作为筛选抗癌药物的基础。
1.3.2 数据处理 收集到的数据先进行数据清洗,去除重复的化合物以及分子指纹或描述符的计算值为0 或无限的化合物,还剩余11 140 种药物,其中抗癌药物200 种,非抗癌药物10 940 种。由于抗癌药与非抗癌药物的数量比值为1∶54.7,说明该数据集非常不均衡,会对结果造成较大的影响,因此需要预先对样本进行处理使其均衡。本文采用两种方法处理数据:第1 种是加权方法,将抗癌药的权重设为54.7,而将非抗癌药的权重设为1;第2 种则是将非抗癌药物分割成55 份,使得每份抗癌药和非抗癌药的比例接近1∶1 的均衡数据集。
1.3.3 特征筛选 由于MACC、PDKit、Mordred 描述符或分子指纹维数较多,计算量大,且可能存在冗余或者相互干扰的特征,导致分类结果不佳,因此需要通过特征筛选,用来提取出与结果相关性强并且相互独立的结构特征,从而筛选出效果较好的分子结构特征组合,提升分类器的分类性能。此外,特征筛选方法还应该保留物理化学意义明确、解释性高的特征,以有利于结合已有的相关化学知识指导新抗癌药物的发现。特征重要性指标有多种形式与计算方法,本文主要采用Pearson 相关系数和卡方检验两种指标。
(1) Pearson 相关系数
Pearson 相关系数(r)广泛地应用于衡量变量之间的线性相关关系,其取值范围是[-1, 1]。当r>0 时,为正相关;当r<0 时,为负相关,且r的绝对值越接近于1,线性相关性越高,其计算式如下:
其中:Xi是所有类别样本中第i个样本对应的该特征变量值;X¯ 是该特征变量中所有样本值的均值;Yi是第i个样本对应的药物类别标签值,其有两种标签值,当第i个样本为抗癌药物时Yi为“1”,为非抗癌药物时Yi为“0”;Y¯ 是所有样本药物类别标签值的均值,式(1)一次只能计算一个特征变量与药物标记之间的关系。
(2) 卡方检验
独立性的卡方检验可用于评估分类变量之间的关联,其中计算单个分类变量较为方便。首先假设分类变量(X)与分类标签(Y)相互独立,则机器学习算法判断的抗癌药物和非抗癌药物个数与实际分类相等,此时卡方计算值( χ2)为0。 χ2越大,说明假设不成立的可能性 ( 1-p,p为分类变量与分类标签相互独立的概率) 越大,即两者关联程度越高。对于X=m类、Y=n类分类问题, χ2和自由度F、p值分别为:

使用Python 工具分别求取与分类结果相关性最大的特征分子指纹或描述符,精简分子特征描述符个数,有助于最终获取数量较少且分类效果较好的描述符组合。
1.3.4 数据分类算法及分类指标 数据分类算法较多,包括决策树、随机森林、支持向量机等算法。它们各有不同的使用范围。由于决策树具有可解释性强[14]、对相关属性处理较好、对初始数据质量要求较低、不需要数据归一化或标准化、可同时处理二元特征和多元特征、运算速度相对较快等优点,且数据集中分子指纹是二元特征、分子描述符是多元特征、非抗癌药物种类多且组成复杂,所以采用决策树为分类算法基础。
(1)决策树参数调整
决策树是一个树状结构,由结点和有向边组成,结点由表示特征的内部结点和表示类别的叶结点组成。根据已知特征属性值和分类结果,可以生成决策树。决策时从根结点出发,根据特征属性值对样本进行分类,可以依据不同标准设定不同阈值进行分类,并判断分类错误率使其最小,分类后根结点分裂,生成子结点,子结点也可以根据新的特征值继续分类,直到分为叶结点。决策树参数调整包括分类标准、决策树深度等。基尼杂质系数(G(P))是常用的分类标准,用以表征错误分类类别的概率,G(P) 越小,分类效果越好,当它为0 时,每个分支均只有一个类别,为完美划分。计算公式如下:
其中,C为分类类别数,p(i) 是一个样本被划分为第i类的概率。
本文将分类标准设为基尼杂质系数,决策树深度调整为3~15 之间,其余均为默认值。
(2)分类性能评估指标
本文使用的数据集标记只有抗癌药物和非抗癌药物之分,因此本文研究的课题实质上是二分类问题。二分类结果预测值和实际标记值如表1 所示。

表1 二分类的结果Table 1 Result of binary classification
分类结果的性能评价指标选择准确率(Acc)、灵敏度(Sen)、精确率(Pre)、F1 分数(F1)、受试者工作特征曲线下(ROC)的面积(AUC) 5 个指标,前4 个指标计算式如下:
其中:NTP、NTN分别为机器学习与实际分类相符合的抗癌药物数、机器学习与实际分类相符合的非抗癌药物数;NFN是被机器学习错分为非抗癌药物但实际标记却是抗癌药物的药物数;NFP是被机器学习错分为抗癌药物但实际标记却是非抗癌药物的药物数。由于本文采用的数据集是根据药物相关数据库和文献来标记,代表实际情况,因此可以通过比较机器学习算法预测的各类药物数以及实际标记的各类药物数的差异,方便直观地体现出机器学习分类效果的好坏。这4 个评价指标都是基于测试集中预测的抗癌或非抗癌药物数与实际抗癌或非抗癌药物数目的差异建立的指标,所以可以评价分类效果性能。
式(6)中Acc计算正确分类的药物总数占测试集中药物总数之比,反映了机器学习算法分类结果的总体准确率。式(7)中Sen计算准确分类的抗癌药物数占实际标记的抗癌药物之比,反映了实际标记抗癌药中机器学习算法能成功识别的抗癌药比例,也就是机器学习对实际抗癌药物的识别程度。式(8)中Pre计算准确分类的抗癌药物数占机器学习预测的抗癌药物之比,反映了机器学习算法预测的抗癌药物中与实际标记相符的抗癌药比例,说明的是预测抗癌药物中有多少是真的抗癌药物。当预测结果与实际标记完全一致时为理想情况,此时NFN=NFP=0,使得Acc=Sen=Pre=1。式(9) 示出了对Sen、Pre进行简单四则运算后得到的综合指标,反映了准确分类的抗癌药物占预测抗癌药和真实抗癌药数目的占比。求偏导可发现Sen、Pre对F1均是促进关系,只有当两者均取最大值1 时它才能取得最大值1。实际情况下,Acc、Sen、Pre、F1这4 个指标越接近1,说明分类越准确。总体而言,这几个指标计算方便、简洁,能较为有效地通过准确分类的抗癌药物的占比来评价分类好坏。
ROC 曲线是根据一系列不同的二分类方式(分界值或决定阈值),以真阳性率(Sen)为纵坐标、假阳性率为横坐标绘制的曲线,是对指标分类结果的可视化表示。相比传统的实验评价方法,ROC 曲线可以根据实际情况,进行多分类的统计分析,AUC的取值范围一般在0.5 和1 之间。AUC数值上等于随机选择的抗癌药物比随机选择的非抗癌药物得分更高的概率[15],AUC越大,分类效果越好。相比于分类误差,它对成本和类别不平衡更不敏感[16],已成为多学科如医疗诊断、药物发现等领域分类性能的评价标准[14,17]。
本文选用的分类评价性指标与文献[7, 18-22]基本一致,具有可靠性。
2 结果与讨论
为了增强结果可靠性,降低分类的随机性并便于比较,本文选用五折交叉验证方法对数据进行训练测试,以测试集的结果来对比经过不同特征处理后数据的分类性能。五折交叉验证是将数据集分成5 份,每次选取1 份作为测试集,其余4 份作为训练集。为了结果的统一性,每次分类结果是经过5 次五折交叉验证后的平均值。
2.1 不采用特征选择
2.1.1 加权数据集 数据集1 是200 个抗肿瘤药与10 940 个非抗肿瘤药组成的数据集,并将抗肿瘤药权重设为54.7,非抗肿瘤药设为1,分别结合不同的分子指纹或描述符,使用决策树在该数据集上实现了抗癌药物的分类。不采用特征选择时不同分子指纹或描述符的分类指标如图1 所示。

图1 不进行特征筛选时加权数据集分类结果Fig.1 Classification result of weighted dataset without feature selection
从图1 分析,不进行特征选择时,RDKit 的5 个分类指标中除Sen外均为最高,但由于它的Sen异常偏低,说明此时RDKit 组成的分类器不能较为准确地筛选出抗癌药物,因为它把接近一半的抗癌药物都分成了非抗癌药物,此时它的识别抗癌药物分类效果较差,Sen低而Acc高说明它只能有效识别非抗癌药物。
对比MACCS 和Mordred,基本上都是MACCS结果高于Mordred 结果,因此MACCS 优于Mordred。又因为MACCS 的Sen在三者中最高,虽然它的Acc不如RDKit,但本文的目的更偏向于从药物中筛选出抗癌药物,因此它的筛选抗癌药物结果为三类描述符中最优。MACCS 其他指标低于RDKit,主要因为其Pre低,也就是划分标准过于宽泛,把较多的非抗癌药物也划分为抗癌药物。
此外还可以发现MACCS、RDKit 与Mordred 的Pre都非常低,主要原因是非抗癌药物远远多于抗癌药物,即便被误认为是抗癌药物的非抗癌药物占非抗癌药物的比例很低,它的绝对数量也远多于正确分类的抗癌药物数。由F1定义可以看出它主要由Sen和Pre中较小的值来确定,由于此处Pre很低,故F1也非常低。
2.1.2 欠采样数据集 数据集2 是将10 940 个非抗癌药物平均分为54 份,每份202 个,由于还有32 个非抗癌药物,随机抽出32 个抗癌药物组成一份小的数据集,每次结果采用这55 份子数据集的平均,重复10 次取平均值,结果如图2 所示。
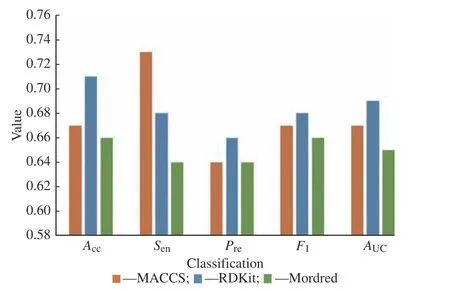
图2 不进行特征筛选时欠采样数据集的分类结果Fig.2 Classification result of under-sampling dataset without feature selection
由图可得,进行欠采样处理后,5 个分类指标数值较为接近,并且除了Acc外,其余指标基本都有提升,特别是Pre和F1值,由于抗癌药物和非抗癌药物个数接近1∶1,因此这两个指标数值均大幅度提升,说明欠采样后模型的不均衡程度显著降低。改用欠采样数据集后Sen也有一定的提升,AUC则变化不大,Acc略有下降。综合来看,有3 个指标上升,1 个指标持平,1 个指标下降,且Acc下降幅度明显小于Pre、F1的上升幅度,说明改用欠采样数据集后模型的分类能力有所提升。
对比3 类分子指纹或描述符分类指标,可以看出Mordred 的分类指标比其余两者要低,说明它的分类效果稍差。对Acc而言RDKit 最优,对Sen而言MACCS 最优,而其他指标RDKit 略优于MACCS,说明单纯看抗癌药物识别度,MACCS 最优,总体而言RDKit 最优。
2.2 采用Pearson 相关系数及卡方检验
采用Pearson 相关系数计算单个描述符或分子指纹对分类的影响。在对前20 个变量描述符之间计算了彼此的相关系数,去掉相关系数大于0.5 的变量,最终按排名分别得到了3 类分子指纹或者描述符中最优的20 个结构描述符组合。采用卡方检验计算各个描述符与分类标签卡方计算值,按照从大到小的顺序排列变量,并用类似方法检验变量之间独立性,去除相关性过高的变量,最后得到最优的20 个结构组合。将两种方法采取的20 个结构描述符结合,筛选出各类分子指纹或描述符中最优的前10 个结构描述符组合如表2 所示。

表2 10 个特征结构分子指纹或描述符Table 2 Ten featured structural fingerprint or descriptors
表2 列出的特征MACCS 列的第1 位代表任意原子与氧形成的双键数是否大于1,从第2 位至第10 位则代表是否存在特定子结构片段,依次代表的子结构片段分别为:任意原子直接与一个硫原子和任意两个原子相连、叔丁基与任意一个原子相连、卤素原子、氮原子与任意除碳和氢之外的原子组成的单键、碳碳双键、硫原子直接与3 个氧原子相连、碳氮双键、含氮杂环、任意六元环(其中有一个为非碳非氢原子)。特征RDKit 列中的第1 位代表由Bertz 提出的分子复杂指数,它基于计算分子拓扑图中各点、各子图以及各种类原子的信息熵总和而得;第2 位和第3 位分别代表苯胺和叔胺个数;第4 位代表0 阶chi( χ 连接价电子指数),由各个骨架原子价电子数目(除去成键的氢原子)为基础计算而得;第5 位代表1 阶chi 连接指数,由骨架上的各对距离为1的原子对的骨架相邻原子数目(除去成键的氢原子) 为基础计算而得;第6 到第10 位分别代表芳香氮、环、亚硝基、甲氧基、吡啶环个数。特征Mordred列的第1~3 位和第5、7 位分别为各对拓扑距离为7、6、5、4、3 的原子对的范德华体积权重下的Moreau-broto 自相关系数,即各个原子对内部原子的范德华体积乘积的加权和;第4、6、8 位为各对拓扑距离分别为7、6、5 的原子对的电离势能权重下的Moreau-broto 自相关系数,即各个原子对内部原子的电离势能乘积的加权和;第9、10 位为各对拓扑距离分别为7、6 的原子对的质量权重下的Moreaubroto 自相关系数,即各个原子对内部原子的质量乘积的加权和。
对比2.1 节中采用不同数据集对药物分类的结果,可以看出,欠采样处理后的药物数据集变得更加均衡,总体而言对Sen、Pre等指标有利,能够更加准确地识别抗癌药物,因此筛选后只选用欠采样数据集,对筛选出来的变量它们的分类结果分别如图3 所示。
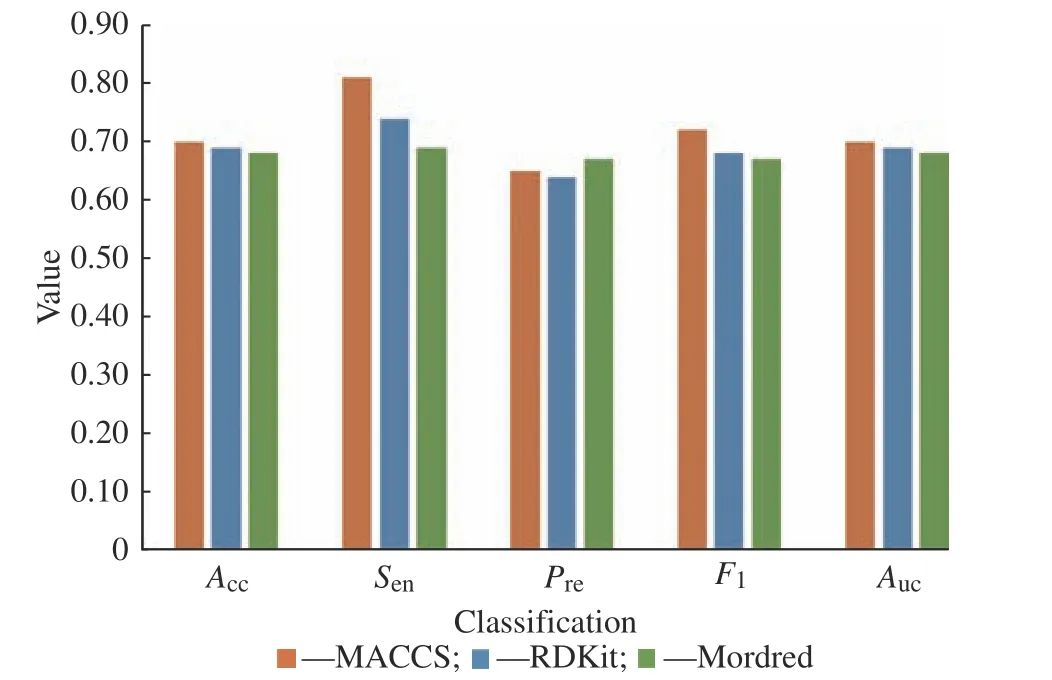
图3 特征筛选后欠采样数据集的分类效果Fig.3 Classification result of under-sampling dataset with feature selection
由图可得,采用Pearson 相关系数及卡方检验进行特征筛选后,3 类分子指纹或描述符的Sen均显著增加,说明特征筛选后分类模型对抗癌药物的准确识别度均明显增加。此外,MACCS 和Mordred 的其他几项指标也在特征筛选后有所增加,说明特征筛选后 MACCS 和 Mordred 的描述符能更好地描述抗癌药物的结构和性质,并明显提高其分类性能。 而对 RDKit 而言,特征筛选后Acc和Pre均有所下降,说明采用的特征筛选方法减低了对非抗癌药物识别能力,但由于抗癌药物识别能力增加,且筛选后F1和AUC基本不变,所以特征筛选仍能基本达成准确分类。
对3 类分子指纹或描述符对比,除Pre外其他指标的大小顺序为:MACCS>RDKit>Mordred,因此在这3 类指纹或者描述符中,MACCS 的分类性能最好,而Mordred 分类性能最差。MACCS 的Sen达到了81%,说明使用它能识别约八成的抗癌药物。综合其他指标可得使用MACCS 的分类模型能准确识别70%的药物。
经过Pearson 相关系数和卡方检验筛选后,MACCS 中10 个指纹结果最好,其中4 个指纹是简单的原子或者共价键,2 个指纹是环状结构片段,其余均为链状片段且基本上都含有支链,同时都有碳、氮、硫、氧等元素。它们都是能相对准确地概括抗癌药物的特征结构组合。综合其他较优的描述符,能准确识别抗癌药物的分子指纹或描述符集合应该满足以下条件:包含基本的官能团如卤素原子、碳碳双键等;包含原子、化学键、链状结构片段、环状结构片段等各种层次的结构;以上结构片段大概率包含碳、氧、氮、硫、卤素原子之中1 种或多种化学元素;除氢外,结构片段原子总数多在2~7 之间。
由MACCS 的Acc(总体药物识别率,70%)低于Sen(抗癌药物识别率,81%),可知MACCS 对非抗癌药物的识别率较低,这有可能是因为非抗癌药物种类远远多于抗癌药物,同时结构多样性,且它的结构片段与抗癌药物的结构片段有交叉,而通过特征筛选后,MACCS 较优的10 个结构指纹组合中,第1、4、6、8 个均为简单的原子或共价键,如卤素原子、碳碳双键等,所以部分非抗癌药物很有可能包含这些简单的原子或共价键,使得对非抗癌药物的识别能力下降。
2.3 结果评价指标的合理性
对比图1 和图2,可以看出,Pre和F1指标能有效区分数据集是否均衡,对于均衡数据集时两者数值均超过0.6;不均衡时数值均较小,接近0。此外,图1、2、3 中所示的Sen指标在各个描述符或分子指纹中差异较大,但均能较为明显地区分出最优值;Acc指标在图1 和图2中能较为明显地区分出最优描述符或分子指纹,所以Acc、Sen、Pre、F1这4 个指标在结果上也能反映出评价指标的合理性。而AUC指标在不同数据集、不同分子描述符之间相差较少,相对而言合理性较Acc和Sen弱,但也能分出最优值。根据分类结果,评价指标基本上均较为合理。
3 结 论
(1)本文采用的欠采样方法可以有效地解决分类数据集极端不平衡的问题。
(2) 两种特征筛选方法的结合使用把原有的分子指纹或描述符精简成了10 个较好的结构分子指纹或描述符组合,能以简短的组合更精准地描述抗癌药物结构,在数据集中有效地筛选出了81%的抗癌药物。
(3) 对现有描述符进行特征选择有助于进一步研究抗癌药物独特性质,有效地筛选抗癌药物,甚至可以指导从结构从头开始设计药物。本研究仅使用了两种特征筛选方法结合决策树建立分类模型,后续可以结合更多特征筛选方法和更多的机器学习算法,深入地研究抗癌药物最为独特的特征属性,更有针对性地识别和筛选抗癌药物。
猜你喜欢
测绘学报(2022年12期)2022-02-13
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
计算机应用与软件(2020年6期)2020-06-16
抗癌之窗(2020年1期)2020-05-21
特别健康(2018年9期)2018-09-26
数字通信世界(2018年1期)2018-04-18
测绘科学与工程(2017年5期)2017-05-07
国外医药(抗生素分册)(2016年3期)2016-07-12
