
药用辅料聚乙二醇6000 中醛类测定方法的比较研究
2024-01-10 11:42石蓉谢莹莹郑金凤张悦尹林高刘雁鸣
药品评价 2023年9期
石蓉,谢莹莹,郑金凤,张悦,尹林高,刘雁鸣
1.湖南省药品检验检测研究院,国家药品监督管理局药用辅料工程技术研究重点实验室,湖南 长沙 410001;2.湖南省药品审核查验中心,湖南 长沙 410001
聚乙二醇6000 是一种无毒、无刺激性的水溶性高分子聚合物,广泛应用于各种药物制剂中[1]。聚乙二醇6000 作为药用辅料主要应用于口服和外用制剂,可作为软膏、栓剂的基质,片剂、胶囊剂的润滑剂,增塑剂等[2]。聚乙二醇6000 现收载于《中国药典》2020 年版四部[3]。目前国内生产企业基本采用乙二醇或二甘醇为起始原料,在碱土金属催化剂的作用下与环氧乙烷聚合生产聚乙二醇6000[4-5]。该聚合物由多个氧乙烯基组成线性链状结构,两端各有1 个羟基,其自氧化过程会发生不同程度的断裂,产生醛类小分子物质,主要为甲醛和乙醛[6-7]。甲醛、乙醛化学性质活泼,可与核酸、蛋白质等发生交联,促进蛋白质羰基化,导致机体应激反应和损伤,从而诱发多种疾病,目前已被国际癌症研究组织认定为1 类致癌物[8]。《中国药典》2020 年版四部标准采用变色酸法对聚乙二醇6000的甲醛含量进行控制。GB/T 14571.3-2008《工业用乙二醇中醛含量的测定-分光光度法》中采用酚试剂法测定乙二醇中总醛含量[9]。现有文献中对低分子醛测定有多种方法,其中2,4 二硝基苯肼(以下称DNPH)柱前衍生-高效液相色谱法应用广泛[10-17]。故本研究通过采用变色酸法测定聚乙二醇6000 中甲醛含量,建立DNPH 柱前衍生-高效液相色谱法测定聚乙二醇6000 中甲醛、乙醛含量,参考GB14571.3-2008 采用酚试剂法测定聚乙二醇6000 中总醛含量这三种方法的比较研究,为药用辅料聚乙二醇6000 醛类测定的标准制修订提供参考和依据。
1 仪器与试药
1.1 仪器
LC-20A 型高效液相色谱仪、PDA 检测器、LCSlution 数据工作站;AUW220D 型电子天平,UV-2550 型紫外分光光度计(均来自岛津有限公司)。
1.2 试药
水中甲醛溶液标准物质(批号为20211208,浓度为10 mg/mL),乙醛对照品(批号为22040034,纯度为99.7%),均购自坛墨质检-标准物质中心。2,4-二硝基苯肼(HPLC 级,批号为C10563969,纯度为98%,购自上海麦克林生化科技有限公司)。乙腈(色谱纯,购自默克公司)。盐酸、硫酸、氨基磺酸、3-甲基-2-苯并噻唑啉酮腙盐酸盐水合物(MBTH)、六水合三氯化铁、变色酸钠均为分析纯。
2 方法与结果
2.1 变色酸法
参照《中国药典》2020 年版四部聚乙二醇6000 的甲醛项[3]测定方法进行试验,在567 nm 波长下测定供试品溶液和对照溶液吸光度,用同法操作的空白溶液进行校正。根据吸光度以外标法计算样品中甲醛含量。
分别制备甲醛对照溶液、乙醛对照溶液进行实验。用空白溶液校正后,结果甲醛对照溶液吸光度为0.468,乙醛对照溶液吸光度为0.002,说明该方法只能测定甲醛,专属性好。
2.2 DNPH 柱前衍生-高效液相色谱法
2.2.1 色谱条件 采用Waters XBridge C18 色谱柱(4.6 mm×250 mm,5 μm),以乙腈-水(60∶40)为流动相等度洗脱20 min,检测波长为360 nm,流速1.0 mL/min,进样体积10 μL,柱温35 ℃。
2.2.2 溶液的制备 衍生化溶液:取经重结晶纯化后的2,4-二硝基苯肼0.25 g,精密称定,置50 mL 棕色量瓶中,加入20 mL 乙腈涡旋使部分溶解,再加入3 mL 盐酸迅速涡旋混匀,超声使溶解,用乙腈稀释至刻度,临用新制。
供试品溶液:取样品0.5 g,精密称定,置10 mL 棕色量瓶中,加入1 mL 乙腈,涡旋使样品溶解后,加入2 mL 衍生化溶液,涡旋混匀,30℃静置15 min,用乙腈稀释至刻度,摇匀,用0.22 μm 有机滤膜滤过,取续滤液作为供试品溶液。
空白溶液:不加样品,按供试品溶液同法操作。
对照溶液:精密称取乙醛对照品0.05 g,置10 mL 量瓶,加乙腈溶解并稀释至刻度,摇匀,即得乙醛贮备液。分别精密量取甲醛标准溶液0.1 mL,乙醛贮备液1 mL,置同一100 mL 量瓶,加乙腈稀释至刻度,摇匀,即得对照混标贮备溶液。精密量取对照混标溶液1 mL,置10 mL 棕色量瓶中,自“加入2 mL 衍生化溶液…”按供试品溶液同法操作。
线性系列标准溶液:分别精密量取甲醛标准溶液0.4 mL,乙醛贮备液4 mL,置同一100 mL 量瓶,加乙腈稀释至刻度,摇匀,即得标准曲线贮备溶液。精密量取标准曲线贮备溶液适量,自“加入2 mL衍生化溶液…”按供试品溶液同法操作,得到线性系列标准工作溶液,详见表1。
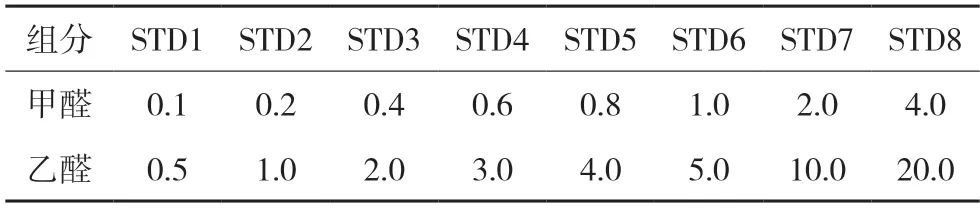
表1 线性系列标准工作溶液 µg/mL
2.2.3 专属性试验 精密量取空白溶液、供试品溶液和对照溶液各10 μL 进样,记录色谱图,详见图1。结果空白溶液对测定无干扰,供试品溶液色谱图中甲醛-DNPH、乙醛-DNPH 峰保留时间与对照品主峰保留时间一致。
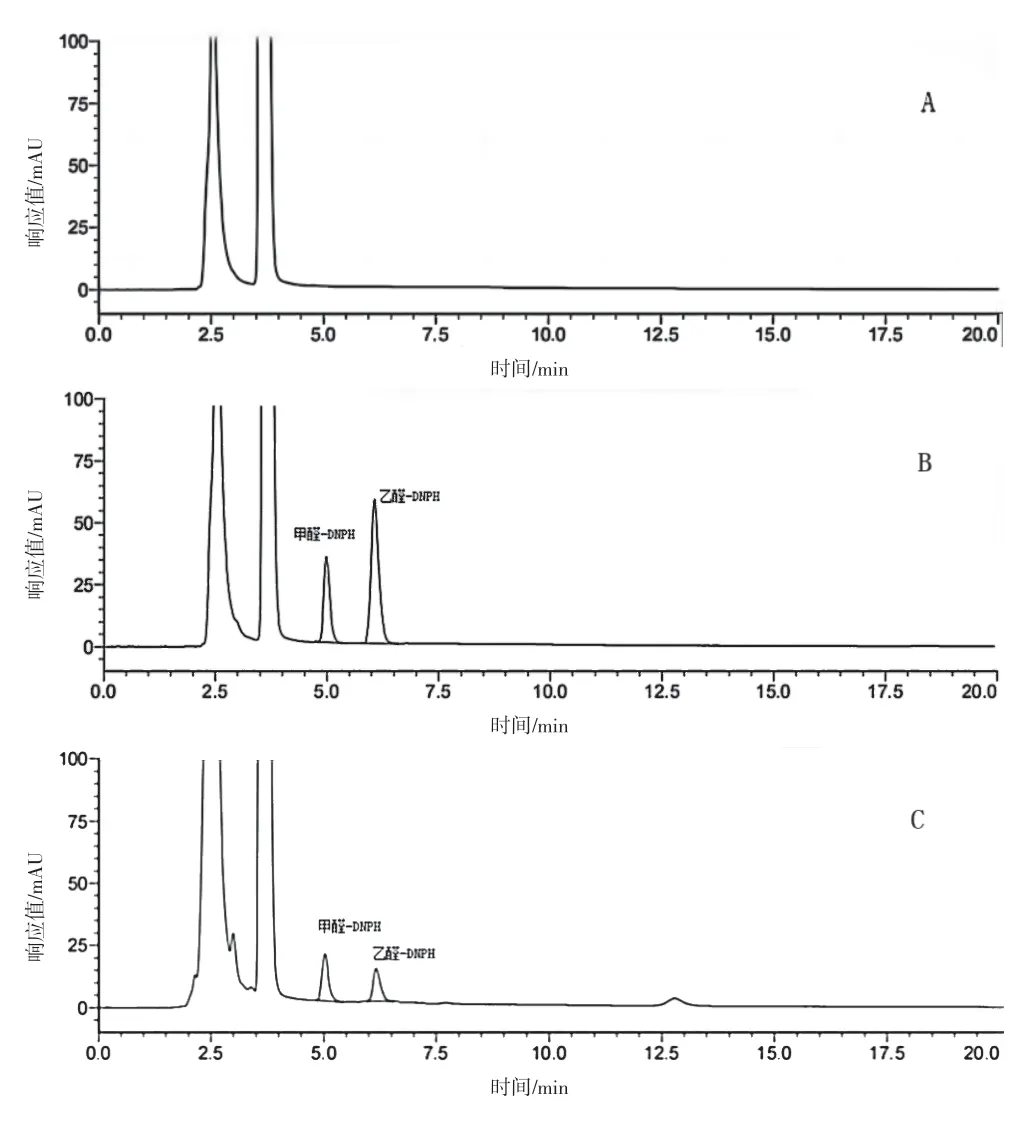
图1 DNPH柱前衍生-高效液相色谱图:A.空白溶液;B.对照品溶液、C.供试品溶液
2.2.4 线性关系、检出限、定量限、精密度与重复性试验 精密量取线性系列标准工作溶液各10 μL 进样,以浓度为X 轴,峰面积为Y 轴,绘制标准曲线。精密量取STD1 溶液1 mL 置10 mL 量瓶,加乙腈稀释至刻度,摇匀,即得检出限溶液。取上述溶液10 μL 进样,分别按S/N=3、S/N=10 计算检测限和定量限。取STD 6 溶液,重复进样6 次,计算峰面积RSD。取同一批样品按“2.2.2”项下方法制备6 份供试品溶液,进行重复性试验,结果详见表2。
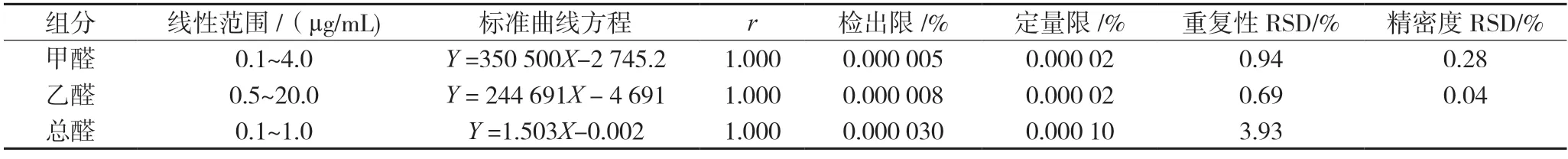
表2 标准曲线、检出限、定量限、重复性与精密度试验结果
2.2.5 稳定性试验 取样品(自编号:J6)和对照贮备溶液适量,按“2.2.2”项下方法制备供试品溶液和对照溶液,于室温放置0、2、4、8 h,分别取样10 μL 测定,以峰面积为考察指标,结果供试品溶液中甲醛-DNPH、乙醛-DNPH 峰面积的RSD 分别为0.86%、0.62%,对照溶液中甲醛-DNPH、乙醛-DNPH 峰面积的RSD 分别为1.55%、0.31%,说明供试品溶液和对照溶液在8 h 内稳定。
2.2.6 回收率试验 分别精密量取甲醛标准溶液1 mL,乙醛贮备液2 mL,置同一100 mL 量瓶,加乙腈稀释至刻度,摇匀,精密量取上述溶液1 mL,置20 mL 量瓶,加乙腈稀释至刻度,摇匀,即得回收标准溶液。取样品(自编号:J6)0.5 g,精密称定,置10 mL 量瓶中,加入1 mL 回收标准溶液,按“2.2.2”项下方法,自“加入2 mL 衍生化溶液…”同供试品溶液操作,平行制备6 份溶液。分别取样10 µL测定并计算回收率,结果甲醛、乙醛平均回收率分别为:98.16%、95.54%,RSD 分别为 2.0%、1.2%。说明方法的准确度良好。
2.3 酚试剂法
2.3.1 测定方法 参照GB/T 14571.3-2008《工业用乙二醇中醛含量的测定-分光光度法》中酚试剂法[9]测定聚乙二醇6000 中总醛。在620 nm 处测定供试品溶液和标准曲线溶液吸光度,用同法操作的空白溶液进行校正,根据吸光度以标准曲线法计算样品中总醛含量。
2.3.2 线性关系、检出限、定量限和重复性试验 取标准曲线系列溶液(0.1 µg/mL、0.2 µg/mL、0.4 µg/mL、0.6µg/mL、0.8 µg/mL、1.0 µg/mL),依法测定吸光度,以浓度为X 轴,吸光度为Y 轴,绘制线性标准曲线。取空白溶液,依法重复测定7 次,计算SD 值。以3 SD/标曲斜率,计算检出限;以10 SD/标曲斜率,计算定量限。取样品(自编号:J15)按供试品溶液同法操作,平行制备6 份供试品溶液,依法测定,结果详见表2。
2.3.3 加标回收试验 取样品(自编号:J15)9 份,各0.2 g,精密称定,分别置50 mL 量瓶中,分为三组,每组3 份,分别加入0.8 mL、1.0 mL、1.2 mL对照贮备溶液,按供试品溶液同法操作,即得加标回收溶液。依法测定,结果低、中、高浓度水平的平均回收率分别为95.09%、99.18%、96.01%,RSD分别为0.66%、0.78%、0.85%,方法准确度良好。
2.4 样品测定
2.4.1 样品来源 共收集到聚乙二醇6000 样品36 批(为国家药品抽检品种,均为药用辅料级),来自7家生产企业,详细信息见表3。
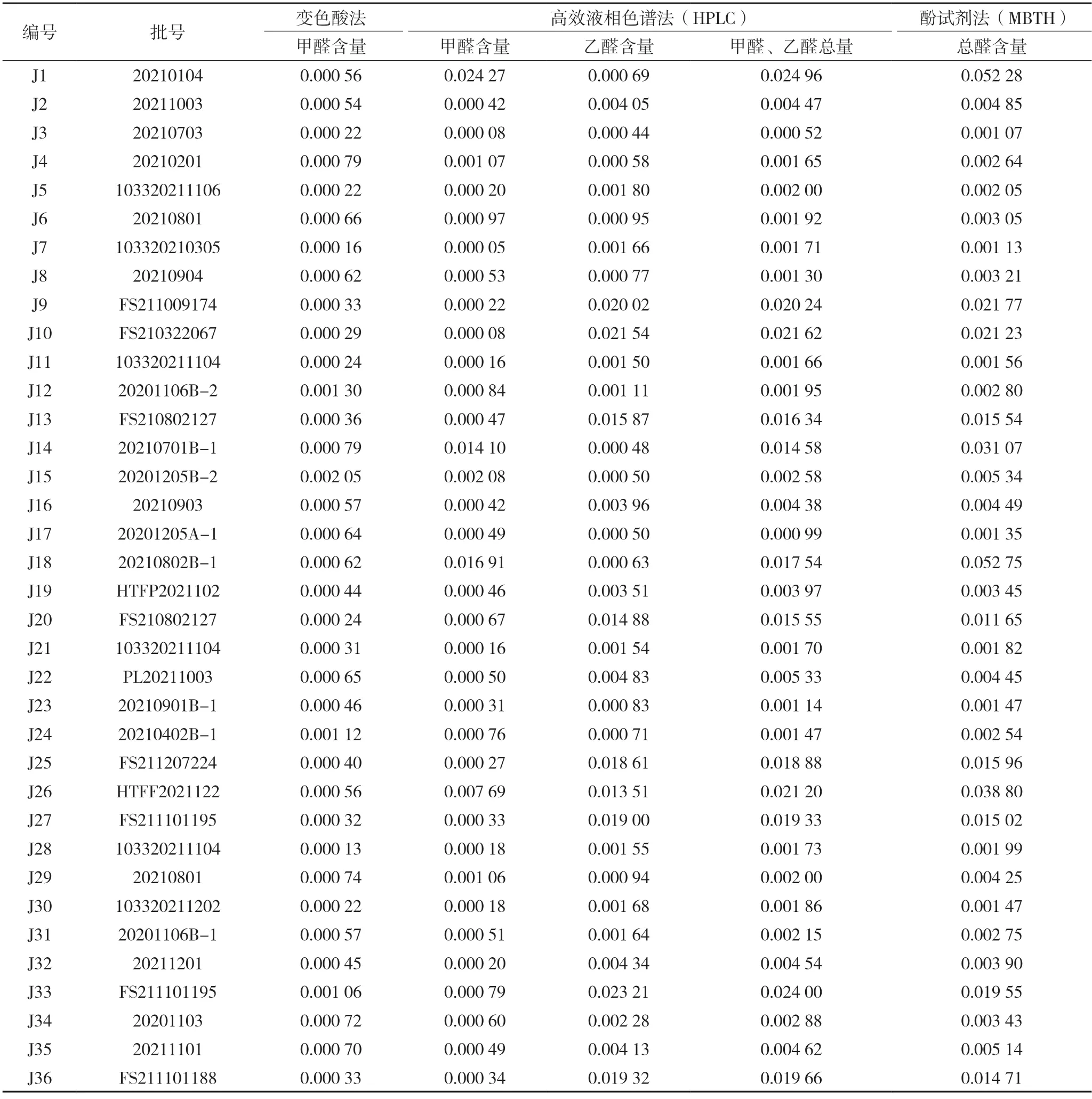
表3 聚乙二醇6000醛含量测定结果比较 %
2.4.2 样品测定结果比较 分别采用上述三种方法,对36 批聚乙二醇6000 样品进行测定,结果比较表详见表3。变色酸法和高效液相色谱法测定甲醛含量比较图见图2,酚试剂法测定总醛含量和高效液相色谱法测定甲醛、乙醛总量比较图见图3。从以上图表可知,按变色酸法测定,36 批样品中甲醛含量为0.000 13%~0.002 05%,均小于法定检验限度(0.003%),符合规定。按高效液相色谱法测定,36 批样品中甲醛含量为0.000 05%~0.024 27%,乙醛含量为0.000 44%~0.023 21%,有4 批样品(编号:J1、J14、J18 和J26)的甲醛测定结果远超法定限度。按酚试剂法测定,36批样品中总醛含量为0.001 07%~ 0.052 75%。
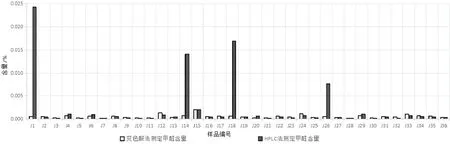
图2 变色酸法和HPLC法测定甲醛含量比较图
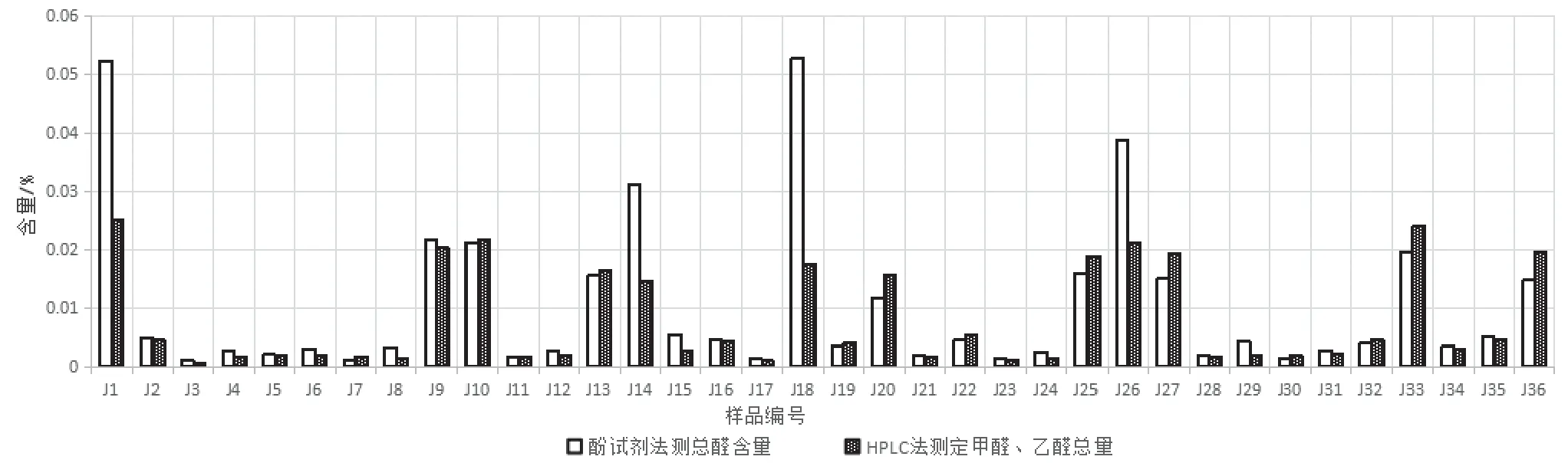
图3 酚试剂法测定总醛含量和高效液相色谱法测定甲醛、乙醛总量比较图
为分析J1、J14、J18(来自A 企业)和J26(来自B 企业)这4 批样品两种方法甲醛测定结果不一致的原因,取样品J1、J26 和J5(来自C 企业,含量小于0.003%)各1 g,精密称定,分别加入1 mL甲醛溶液(30 μg/mL),按变色酸法制备样品加标回收溶液,在400~700 nm 紫外波长范围进行扫描。结果显示,样品J1、J26 均对标准溶液的显色有干扰,样品加标溶液在567 nm 处无最大吸收,而来自C 企业的J5 样品对标准溶液的显色无干扰,样品加标溶液在567 nm 处有最大吸收。
3 讨论
3.1 衍生化条件考察
采用DNPH 柱前衍生法进行样品处理,关键在于衍生液和衍生化过程。有文献[16]报道2,4-二硝基苯肼本身会带来甲醛、乙醛干扰。本研究对购买的HPLC 级2,4-二硝基苯肼进行重结晶纯化(称取2,4-二硝基苯肼5 g,置圆底烧瓶中,加入100 mL 乙腈,90 ℃水浴加热60 min 使溶解,60℃水浴中结晶,直到得到较大结晶,弃去上清液,用乙腈清洗结晶3 次,105 ℃干燥,即得),经过空白试验验证,可排除醛类干扰。同时,衍生液应临用新制,比较配制后1 h 和8 h 的衍生液处理样品和对照溶液。样品中的甲醛、乙醛含量分别下降12.20%和11.14%,对照溶液中的甲醛、乙醛含量分别下降7.05%和8.21%。衍生温度对衍生结果也有一定影响,分别考察30 ℃、40 ℃和50 ℃三个温度梯度,结果显示同一反应时长下,随着温度增高,对照溶液中甲醛、乙醛含量呈下降趋势。
3.2 三种醛类测定方法比较
本研究对聚乙二醇6000 中醛类测定方法进行了比较,结果表明除J1、J14、J18 和J26 这4 批样品外,变色酸法与高效液相色谱法测定的甲醛含量,高效液相色谱法测定的甲醛、乙醛总量和酚试剂法测定的总醛含量基本一致。
变色酸法是法定标准,该方法中变色酸和甲醛以浓硫酸为介质,生成紫色络合物,专属性好,但是该方法易受样品基质干扰,当样品中含有干扰显色物质时,比如其他醛类或杂质,样品在检测波长处无最大吸收,造成甲醛含量测定结果远低于其实际含量。本研究建立的DNPH 柱前衍生HPLC 法简便易操作,方法学验证结果表明方法可行,可用于聚乙二醇6000 中甲醛、乙醛含量测定。酚试剂法测定总醛含量的方法学验证结果符合要求,4 批样品(J1、J14、J18 和J26)总醛测定结果均高于HPLC 法测定甲醛、乙醛总量。推测可能是该4 批样品中含有其他醛类,导致测定结果偏高。故三种醛类测定方法中,通过HPLC 测定聚乙二醇6000中甲醛、乙醛含量不受样品干扰,测定结果更为准确,可以作为甲醛、乙醛定量分析方法,结合酚试剂测定总醛含量,可以从总体上控制药用辅料聚乙二醇6000 的醛类物质。
猜你喜欢
煤炭与化工(2023年8期)2023-10-11
山东化工(2018年15期)2018-09-20
深圳职业技术学院学报(2018年1期)2018-01-27
首都食品与医药(2015年18期)2015-11-03
中国当代医药(2015年8期)2015-03-01
中国塑料(2014年1期)2014-10-17
中学教学参考·理科版(2014年4期)2014-08-21
癌变·畸变·突变(2014年2期)2014-03-01
食品科学(2013年13期)2013-03-11
食品科学(2013年13期)2013-03-11
