
2013-2022年江苏省柯萨奇病毒A6型全基因组序列特征分析
2024-01-15 12:23鲍倡俊朱立国胡建利
中国人兽共患病学报 2023年12期
樊 欢,鲍倡俊,朱立国,胡建利,嵇 红
手足口病(Hand,foot and mouth disease,HFMD)是一种常见儿童传染病,由多种类型人肠道病毒(EV)引起,多发于低龄婴幼儿童,临床表现为发热、手、足、口和臀等部位扁平状红色疱疹[1-3];该病属于自限性疾病,绝大多数病例病情轻微可在1~2周内自愈,仅少数患儿病情危重,病情发展迅速,累及神经系统和循环系统,出现心肌炎、脑炎、肺水肿和弛缓性麻痹等并发症,甚至导致死亡[4-5]。
人肠病毒属于小核糖核酸病毒科肠道病毒属,是一种无包膜、单股正链RNA病毒[6];目前已知的人肠道病毒的血清型超过100多种,根据人肠道病毒VP1 核酸序列的差异进行分类,将人肠道病毒分为A、B、C和D 4组。其中,肠道病毒A组在手足口病的发病中起着极其重要的作用,主要由20多种血清型组成,包括柯萨奇病毒1-10型、12型、14型、16型、21型和EV71型等[6-8]。往年手足口病多由肠道病毒71型(EV-A71)和柯萨奇病毒A16型(CV-A16)引起[9]。然而,由于疫苗的应用和病毒本身流行的特征,近年来HFMD的主要病原体发生了变化,非EV-A71和非CV-A16感染占比增加,其中柯萨奇病毒A6(CV-A6)逐渐占据国内手足口病原谱的主导地位[10-11]。CV-A6感染所致的临床症状与典型的手足口病症状稍有不同,易发高热,抽搐,其皮疹可分布全身,如四肢、躯干、臀部和面部等部位,形态多样,且皮疹面积较大,持续时间较长,皮疹消退后可出现脱皮[12-13],部分病例痊愈后还会出现脱甲症,严重者可累及循环系统和神经系统[14]。此外,CV-A6还是疱疹性咽峡炎的主要病原体[15],然而该病并没有被纳入中国疾病监测网系统,因此,CV-A6引起的疾病负担可能被严重低估。
本研究收集了2013-2022年江苏省手足口及疱疹性咽峡炎患者CV-A6阳性标本,对2013-2022年期间分离到的35份CV-A6毒株进行全基因组测序,旨在了解和掌握江苏省CV-A6病原优势株的基因型、进化亲缘关系、基因重组及重要氨基酸位点突变情况,为今后的CV-A6感染的防治工作提供基础资料。
1 材料与方法
1.1 材 料
1.1.1 资料和标本来源 本研究通过江苏省手足口病监测哨点收集了2013-2022年CV-A6 阳性标本的咽拭子或肛拭子;通过“中国疾病预防控制信息系统”收集标本的病例类型及病原学检测结果等。
1.1.2 仪器与试剂 全自动核酸提取仪及其配套试剂购自天隆科技有限公司,ULSEN超灵敏度肠道病毒全基因组捕获试剂盒A组购自北京微未来科技有限公司,Iseq100 二代测序仪及Nextera XT DNA Library Preparation Kit购自Illumina公司,Opti-MEM培养液、胎牛血清和青霉素购自GIBCO公司。
1.2 实验方法
1.2.1 病毒分离 人横纹肌肉瘤(RD)细胞接种于细胞培养板,37 ℃、5% CO2培养箱培养,待细胞生长至80%左右融合度时,将CV-A6阳性的标本过滤后接种于RD细胞进行病毒分离,观察细胞病变(cytopathic effect,CPE)情况,7 d内无细胞病变,则再次盲传,盲传2代,若出现细胞病变,反复冻融3次后收集培养液,并储存于-80 ℃。
1.2.2 核酸提取 采用天隆科技公司的全自动核酸提取仪及其配套试剂按照其说明书操作步骤提取CV-A6毒株核酸,收集的核酸置于-80 ℃保存。
1.2.3 文库制备及上机测序 参照ULSEN超灵敏度肠道病毒全基因组捕获试剂盒A组说明书配置反应体系,对CV-A6毒株核酸进行全基因组逆转录后,PCR扩增富集,扩增产物采用Nextera XT DNA Library Preparation Kit进行文库制备,变性和稀释好的文库使用Illumina Iseq100平台二代测序。利用CLC Genomics Workbench 21软件对原始数据进行过滤,与CV-A6参考序列进行比对和拼接。
1.2.4 生物信息学分析 自GenBank中下载CV-A6不同基因型代表株序列作为参考序列,采用DNASTAR软件对获得的CV-A6毒株全基因组序列与CV-A6参考序列进行核苷酸和氨基酸序列分析及相似性比较,采用Mega7.0软件的邻接法(Neighbor-Joining)构建CV-A6 VP1和3D区段序列的系统进化树[16-17],自展值设置为1 000。对CV-A6毒株P1区和3D区氨基酸序列变异位点进行统计分析,统计学分析采用Excel 2007软件进行数据整理。
1.2.5 重组分析 采用RDP4.0软件的X-Over模块,选择默认参数进行重组分析。采用SimPlot 3.5.1软件进行相似性分析。
2 结 果
2.1 测序结果分析 通过病毒分离获得2013-2022年江苏省共35株CV-A6毒株,其中手足口轻症病例来源22株,手足口重症病例来源7株,疱疹性咽峡炎轻症病例6株。这35株CV-A6分离株来源信息如表1所示。对35份毒株进行全基因组测序并获得全基因组序列。这35株CV-A6毒株中,14株基因组长度为7 434 bp,5′-UTR为747 bp; 10株基因组长度为7 433 bp,5′-UTR为746 bp; 13株基因组长度为7 428 bp,5′-UTR为741 bp; 2株基因组长度为7 412 bp,5′-UTR为725 bp; 1株基因组长度为7 420 bp,5′-UTR为733 bp;这35株CV-A6在编码区没有碱基的缺失与插入,但在3′-UTR最末端的7 432-7 434位点有5株存在3个碱基缺失。

表1 2013-2022年江苏省CV-A6分离株样本信息
2.2 相似性分析 35株CV-A6毒株的全基因组核苷酸和氨基酸序列相似性分别为87.5%~99.6%和97.0%~99.8%,这35株毒株的核苷酸序列差异较大,但对应的氨基酸序列差异性无明显变化。与CV-A6原型株Gdula/USA/1949的全基因组核苷酸序列相似性仅为80.3%~81%,氨基酸序列相似性为94.7%~95.3%。各区段比对发现,5′-UTR、P1、P2、P3各个区域核苷酸相似性分别为94.7%~100%、88.1%~100%、87.1%~100%和84%~100%;编码P1、P2、P3各区域氨基酸相似性分别为97.1%~100%、96.7%~100%和95.5%~100%;VP1区核苷酸和氨基酸相似性分别为87.7%~100%和97.4%~100%。
2.3 CV-A6分离株VP1基因进化分析 选取国内外其他有代表性的CV-A6 VP1全长序列,与2013-2022年35株江苏省CV-A6毒株进行遗传进化分析,并构建系统进化树。结果表明,其中34株CV-A6毒株与D3a基因亚型代表株处于同一分支上,1株毒株Xinzixuan(Nanjing/2014)与基因D2亚型代表株处于同一分支(图1)。这34株D3a亚型的CV-A6毒株相互之间的遗传距离较为集中,亲缘关系较近,相对国外地区的毒株序列,与国内地区(如山东、云南、广东、浙江等省份)毒株的遗传距离和亲缘关系更为接近。同时,进化树显示,这34株毒株在进化树上呈现地区分布的聚集性,但轻症和重症来源的毒株、不同临床症状来源的毒株(疱疹性咽峡炎病例与手足口病例)在进化树上呈现随机分布。

●:轻症HFMD CV-A6 毒株;●:重症HFMD CV-A6 分离株;●:疱疹性咽峡炎CV-A6 分离株
2.4 CV-A6毒株3D区进化重组分析 基于3D区进化树的的不同分支划分了不同重组模式,本研究发现,2013-2022年江苏省流行的CV-A6出现过4个重组进化分支,其中D3a基因亚型包含3个重组进化分支。其中26株CV-A6毒株与RF-A型代表株处于同一分支上,5株CV-A6毒株与RF-L型代表株处于同一分支,3株CV-A6毒株与RF-K型代表株处于同一分支,此外,D2基因亚型的毒株Xinzixuan(Nanjing/2014)与C型代表株处于同一分支(图2)。
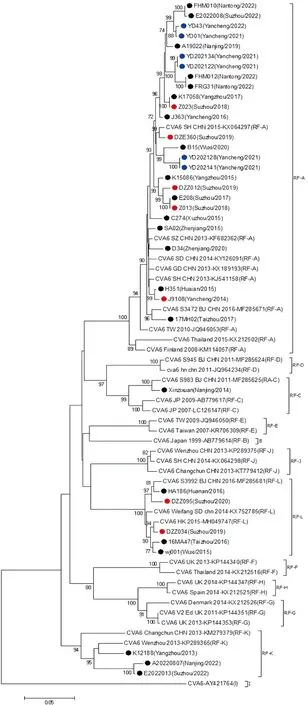
●:轻症HFMD CV-A6 毒株;●:重症HFMD CV-A6 分离株;●:疱疹性咽峡炎CV-A6 分离株
2.5 重组分析 通过RDP4.0软件对35株CV-A6毒株与EV-A组各原型株或代表株全基因组序列进行重组分析,结果显示:毒株K12188(Yangzhou/2013)、DZZ095(Suzhou/2020)、wj001(Wuxi/2015)、J9108(Yancheng/2014)和16MA47(Taizhou/2016)等均具有重组信号,亲本序列有CVA14/PZ15G/JS/2012-KP036483、CVA8/SZ124/CHN/2012-KM609476、CVA7-AY421765和CVA16-U05876等。
为进一步验证重组分析结果的可靠性,运用SimPlot 3.5.1软件对上述具有重组信号的江苏省毒株序列、相应亲本序列及其他EV-A原型株序列进行相似性分析,结果如图4所示:江苏省CV-A6毒株与CV-A6原型株Gdula/USA/1949在结构蛋白序列(P1区)上具有较高的相似性。而在非结构蛋白(P2、P3区)、5′-UTR和3′-UTR区,江苏省CV-A6毒株与其他EV-A序列相似性更高。例如BootScan分析结果也表明:RF-K型代表株K12188(Yangzhou/2013)在5′-UTR、部分P2C区段、P3区和3′-UTR区与CVA14/PZ15G/JS/2012-KP036483相似性较高;RF-L型代表株DZZ095(Suzhou/2020)、wj001(Wuxi/2015)和16MA47(Taizhou/2016)在3D区和3′-UTR区与CVA8/SZ124/CHN/2012-KM609476相似性较高;上述结果提示江苏省CV-A6毒株与其他EV-A病毒具有多重重组现象。
2.6 江苏省CV-A6毒株主要氨基酸变异位点分析 对CV-A6毒株P1区氨基酸序列变异位点分析发现,如表2所示,与原型株Gdula/USA/1949相比,江苏省CV-A6分离株在保守序列P1区中仍然存在较为频繁的点突变,这些突变在轻症和重症病例中呈现无规律散在分布,并未呈现年度和地区规律性的氨基酸突变。此外,值得注意的是,在VP2区第179 aa位点中,RF-C型毒株发生了V→I的突变;而在VP2区第180 aa位点,除RF-C型毒株外其他均发生了F→Y的突变。
对江苏省CV-A6分离株3D区氨基酸序列变异位点分析发现,结果如表3所示:与原型株Gdula/USA/1949相比,不同重组模式的CV-A6分离株氨基酸突变位点存在差异,除此之外,江苏省近2年的CV-A6毒株在3D区166 aa位点更易发生I→V的突变,在370 aa位点更易发生E→G的突变。

表3 江苏省CV-A6分离株3D区氨基酸变异位点分析
3 讨 论
HFMD为全国广泛流行的儿童传染性疾病,江苏省气候温暖潮湿较北方地区发病率高[3],引起较多的重症和死亡病例。以往病原学监测数据显示,2009-2017年江苏省引起HFMD的主要病原为EV71和CVA16[18-19],之后EV71感染率呈现下降趋势,CV-A6感染率逐步上升,呈现流行趋势。CV-A6被划分为A、B、C、D四个基因型,世界流行的CV-A6绝大部分属于D基因型,尤其是D3亚型,提示D3基因亚型毒株可能是引起CV-A6在世界范围内流行的主要病原[20-23]。中国流行株只见于D2和D3亚型,D3亚型是中国大陆2008年以后的绝对优势基因亚型,尤其是D3a分支,所以这几年属于CV-A6 D3a亚型的暴发阶段[11]。本研究基于2013-2022年CV-A6江苏省毒株全基因组序列,对各个编码区进行遗传进化及相似性分析,为江苏省HFMD防控策略的制定及CV-A6的流行趋势提供了详细的分子病原学数据。
2013-2022年江苏省CV-A6毒株相似性分析结果显示:基于CV-A6全长序列,其核苷酸序列相似性明显低于氨基酸序列相似性,推断CV-A6在进化过程中的点突变大多为同义突变。此外,各区段比对发现,非结构蛋白P2和P3区核苷酸序列相似性低于P1区序列核苷酸相似性,这可能是由于CV-A6在流行传播过程中,与其他EV-A在P2区和P3区发生不同程度的重组造成的。
基于VP1序列的系统进化树显示,2013-2022年35株CV-A6江苏省分离毒株与原型株Gdula/USA/1949及国外地区毒株遗传距离较远,亲缘关系远,与国内地区如云南、北京、广州等地分离株的遗传距离较近,亲缘关系近。本研究中的34株CV-A6分属于D3a亚型,只有1株Xinzixuan(Nanjing/2014)分属于D2亚型;VP1进化树中也可以看出在2014年之前存在D2和D3亚型共存的情况,而2014年之后全部转为D3a亚型,这可能与D3a亚型相对其他亚型具有更强的传播性和感染性有关。上述结果和其他省市如北京、深圳等地区的CV-A6流行情况一致[22,24-26],提示各地区 CV-A6通过多种传播链从而共循环同进化。此外,轻症和重症病例、疱疹性咽峡炎和手足口病例在进化树上呈现随机分布,提示CV-A6引起的临床差异可能与VP1区的基因序列无关。
研究表明,非结构蛋白在病毒复制中起着重要作用,CV-A6可能通过与其他肠道病毒的非结构蛋白重组,更有利于病毒在人体内的复制和传播,因此,基于CV-A6 3D区进化树的不同分支划分了12种不同重组模式(RF):RF-A到RF-L[22,24]。本研究分离到的CV-A6毒株出现了4个重组模式,其中34株D3a基因亚型分离株存在3个重组模式:RF-A、L和K,且以RF-A为主。D2基因亚型的Xinzixuan(Nanjing/2014)则为RF-C重组模式,上述结果提示过去10年里,江苏省CV-A6与其他A组肠道病毒有基因重组的可能。
进一步重组分析结果显示,本研究有多株CV-A6分离株与CV-A6原型株在P1区域高度匹配,但在非结构蛋白(P2、P3)、5′-UTR和3′-UTR区发现与其他EV-A存在重组,且重组方式存在多样性。已有文献报道,CV-A6可通过与其他EV-A的重组向更有利于病毒在人体内复制和传播的方向进化。例如,近年来,不同重组模式的CVA6先后在上海、南京、香港引起手足口病的持续暴发[10,27-28],此外,2016-2017年在澳大利亚流行的CV-A6与同期流行的CV-A4也有发生重组[29]。综上所述,本研究发现江苏省传播的CV-A6通过重组的方式不断进化,具有成为高致病力病毒的危险,而近年来CV-A6持续流行是否与现有CV-A6流行株发生重组有关还需更多实验数据来证实。
CV-A6在传播过程中,衣壳蛋白P1区中的大部分密码子都发生了强负选择,其中正向选择位点VP3180和VP130可能在适应新宿主的过程中发挥重要作用;而位点VP2130、VP2172、VP2179和VP2180位于表位EF环,突变位点VP1242位于表位HI环,这5个位点的变化可能会优化对宿主的适应性,对近年来CV-A6的高致病性和传播性上发挥了重要作用;此外,有文献报道VP2244、VP15、VP17、VP130、VP1137、VP1174和VP1242位点的突变可能会使得衣壳蛋白的结构、潜在的受体结合位点发生一些难以察觉的变化[23-24]。本研究对江苏省35株CV-A6分离株氨基酸位点进行分析,与原型株Gdula/USA/1949相比,大部分突变位点在不同分离株中不存在规律性分布。但是在VP2区第179 aa位点,只有RF-C型分离株存在V→I的突变;而VP2区第180 aa位点,除RF-C型分离株外其他均发生了F→Y的突变。
由于3Dpol具有谱系特异性,3D区RdRp蛋白氨基酸的突变可能会最终影响CV-A6的致病性[24]。已有文献报道肠道病毒3D区存在毒力位点,3D区的突变或重组可能会对临床表型产生影响[30-31]。本研究中,与原型株Gdula/USA/1949相比,江苏省不同重组模式的CV-A6分离株3D区氨基酸序列变异情况具有明显的差异,其中,江苏省近两年的CV-A6分离株3D区在166 aa位点更易发生I→V的突变,在370 aa位点更易发生E→G的突变。上述3D区位点的突变是否对近年CV-A6 的传播性和致病性产生影响还需进一步研究。
综上所述,本研究通过对2013-2022年江苏省CV-A6血清型全基因组测序分析,有助于了解江苏省CV-A6基因重组及遗传进化关系,解释了近年来CV-A6成为手足口病的主要病原体的可能原因,提示江苏省流行的CV-A6在全基因组层面有明显的遗传多样性,为江苏省CV-A6感染防控策略和疫苗研制提供一定基础数据。
利益冲突:无
猜你喜欢
数学物理学报(2022年5期)2022-10-09
科学大观园(2022年2期)2022-01-23
河北画报(2020年8期)2020-10-27
中国医药指南(2017年3期)2017-11-13
安徽医科大学学报(2016年12期)2017-01-15
浙江大学学报(工学版)(2016年2期)2016-06-05
动物医学进展(2015年10期)2015-12-07
现代检验医学杂志(2015年6期)2015-02-06
特产研究(2014年4期)2014-04-10
郑州大学学报(理学版)(2014年3期)2014-03-01
