
第一性原理研究Hf对ZrCoH3放氢的影响
2024-01-18 08:38杨友山
原子与分子物理学报 2024年4期
曾 祥,杨友山
(铜仁学院 农林工程与规划学院,铜仁 554300)
1 引 言
氢同位素储存与供给系统(SDS)是磁约束聚变堆氚燃料循环系统的重要组成部分[1].SDS中储氢材料可以分为金属、非金属和有机液体三类[2].与非金属储氢材料和有机液体储氢材料相比,氢的化学性质很活泼,氢气能与储氢金属或合金发生反应,形成金属氢化物ß相或者固溶在金属中形成α相,在改变外部压力或者改变外界环境温度的条件下,便能解吸附金属氢化物中氢,基于这些特性,金属储氢被认为是一种安全可靠的储氢方法[3,4].
近年来,由于ZrCo在国际热核试验堆项目(ITER)中氢同位素的储存、供应、回收方面具有良好的特性[5,6],ZrCo被认为是ITER研发团队选取为用于氢同位素快速储存与供给的重点备选材料[7-10].但是ZrCo氢致歧化生成稳定的ZrH2相和不吸氢ZrCo2相[7-9](2ZrCoH3→ZrCo2+ ZrH2+2H2,2ZrCo+H2→ZrCo2+ ZrH2),永久性降低ZrCo储氢量和循环稳定性,成为其在涉氚体系中最大的障碍[11].为了改善ZrCo氢致歧化,提高ZrCo抗歧化性能,掺杂被证实是一种有效方式.例如:利用电弧熔炉方法制备Ti掺杂改性的ZrCo,采用恒容等温法测试ZrCo及其氢化物的歧化动力学,发现Ti掺杂后的相结构表征为单一固溶体相,材料吸氢/放氢性能测试表明放氢坪台压力随着Ti含量的增加而上升,掺杂后的新合金在达到100 KPa放氢坪台压的温度为600 K左右,在该温度下放氢可以避免歧化反应的发生[12].通过电弧熔炼方法研究了Ti元素掺杂ZrCo合金的比例对合金储氢性能和结构的影响,采用Sievert-型容量仪器测量吸放氢性能和X射线衍射(XRD)材料表征技术表征放氢产物(ZrCo2和ZrH2),发现Ti掺杂不改变ZrCo合金的晶体结构,并且能够有效地抑制合金歧化反应的吸附平衡压力,在新形成的三种含Ti合金中,Zr0.8Ti0.2Co合金的反歧化性能最好,有利于氢及同位素的储存[13].掺杂后ZrCo氢致歧化得到改善,主要归因于掺杂后ZrCo晶胞内供氢原子占据的间隙位尺寸发生变化.显然,众多学者利用掺杂手段有效的改善了ZrCo抗歧化性能,提高了ZrCo合金的脱氢性能,然而,现有的ZrCo体系仍无法满足ITER项目实际需要,为了开发实用化的ZrCo-H体系,需要深入研究ZrCo储氢的掺杂作用机制.
最近,Nakamori等[14-16]通过对系列单离子金属硼氢化合物M(BH4)n的热力学性能进行研究发现,金属Mg或金属Mg化合物掺杂LiBH4制备双离子硼氢化合物LiM(BH4)n,成功实现LiBH4结构“失稳”从而提高了LiBH4储氢能力[17,18].Yang等[19]研究了掺杂元素对ZrCo氢化/脱氢性能的影响,对Hf单独替代Zr后的ZrCoH3进行了Bader分析,发现Zr与其替代元素转移到H的电子多于从Co及其替代元素转移的电子,表明H与Zr及其替代元素之间的结合表现出强的离子和弱共价特性,H与Co及其替代元素之间的结合是强共价和弱的离子特性,电荷密度分布和态密度的分析结果与电负性完全吻合,证实了Zr原子和Co原子与氢原子之间的成键性质不同,弱化了ZrCoH3结构的稳定性.然而,对于Hf掺杂带来的ZrCoH3结构 “失稳”,放氢能力的提高的Hf作用机制,Yang等没有深入报道,且目前相关文献也较少.鉴于此,为了进一步提高ZrCoH3的储氢能力,了解掺杂ZrCoH3的电子结构机制,这里,Hf被设计为通过占据Zr原子、Co原子和间隙位点来添加到ZrCoH3中,本工作采用基于第一性原理密度泛函理论计算研究ZrCoH3掺杂的Hf原子占位能、氢的离解能、电子结构以及无Hf和掺杂Hf的ZrCoH3的键合特性,旨在深入了解ZrCoH3-Hf体系的放氢机理.
2 计算细节
本研究中第一性原理计算采用Materials Studio软件中的Cambridge Serial Total Energy Package(CASTEP)[20]模块,CASTEP模块是基于平面波赝势方法和密度泛函理论完成的,即用赝势代替粒子势.计算中,选择广义梯度近似下Perdew-Wang91泛函(GGA-PW91)描述交换关联能,势函数采用倒空间表达的超软势(Ultrasoft)赝势,选择超软赝势描述离子实与价电子间相互作用[21].平面波截止能为350eV,布里渊区K点网格数为2×3×2.各原子外层电子的电子组态分别为:Zr:4s24p64d25s2、Co:3d74s2、Ti:3s23p6d24s2、Hf:5d26s2、H:1s1.采用Broyden-Fletcher-Goldfarb-Shanno (BFGS)方法对晶胞进行几何优化,以求得局域最稳定结构[22].几何优化结束时,体系总能量收敛值为2×10-6eV/atom,公差偏移小于0.002Å,应力偏差小于0.02 GPa.利用优化的晶体模型进行体系的单点能及电子结构计算.
ZrCoH3空间群属于Cmcm,为正斜方晶结构类型,晶格常数分别为a=3.53 Å,b=10.48 Å,c=4.3 Å[23].对ZrCoH3晶胞进行几何结构优化,优化后晶格常数如表1所示,与实验值[24]及理论值[23]非常吻合.将ZrCoH3晶胞沿着a轴和c轴延伸建立2×1×2超胞模型如图1所示,其中H1、H2为晶胞中的两类氢原子,S1、S2分别代表被取代的Zr、Co原子,S3为间隙掺杂位置.该模型包含16个Zr原子、16个Co原子、48个H原子,此体系记为M0.对ZrCoH3-Hf掺杂体系的晶胞结构进行几何优化,所得结果列于表1,很显然与本体系M0晶胞晶格常数相比,Hf掺杂使体系晶胞略沿a轴、c轴延伸,沿a轴收缩,这源于掺杂元素Hf的原子半径与被掺元素Zr、Co存在差异.单质Zr、Co、Hf原子的能量分别为:-1281eV、-1042.9eV、-408.8eV.为了计算H2能量,可以利用第一性原理计算得到E(H2)=-31.79 eV,该结果与文献[25,26]的报道E(H2)=-31.292 eV相吻合.

表1 Zr16-xCo16-yHfx+y+zH48体系的晶胞参数及能量计算值
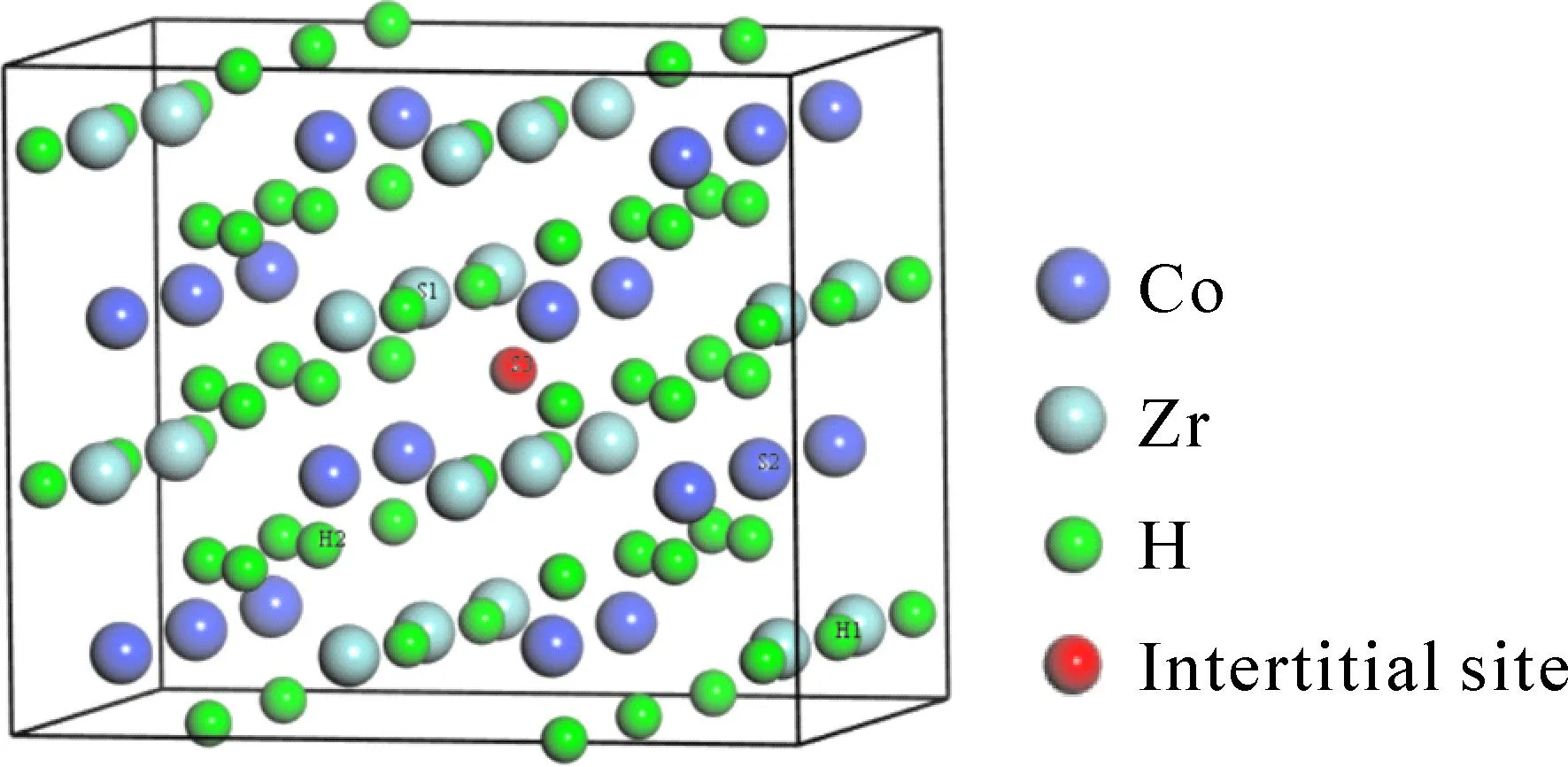
图1 ZrCoH3的2×1×2超胞模型
3 结果与讨论
3.1 位置能
本文中的研究体系记为Zr16-xCo16-yHfx+y+zH48,其中(x=0,y=0,z=0)代表纯Zr16Co16H48化合物(M0);(x=1,y=0,z=0),(x=0,y=1,z=0)和 (x=0,y=0,z=1)表示Hf占据Zr原子位点,Co原子位置,和间隙位点,分别产生Co-、Zr-和间隙位点取代化合物,即Zr15Co16HfH48(M1)、Zr16Co15HfH48(M2)和Zr16Co16HfH48(M3).所研究系统中的Hf浓度约为7.0at%.M0、M1、M2和M3都完全弛豫以获得优化的晶格参数(Lop)如表1所示.由于Hf掺杂,晶胞体积发生膨胀,为H原子扩散提供了更多的点位.
研究Hf原子部分取代Zr16Co16H48体内Zr、Co原子所消耗能量,即在Zr16Co16H48体系中Hf原子掺杂的位点偏好可以用选择能Eocc估计,定义如公式(1)[27]:
Eocc=E(Zr16-xCo16-yHfx+y+zH48)-
E(Zr16Co16H48)-[(x+y+z)E(Hf)-
xE(Zr)-yE(Co)]
(1)
式中x、y、z与上述定义相同;E(Zr16-xCo16-yHfx+y+zH48)、E(Zr16Co16H48)、E(Hf)、E(Zr)、E(Co)是相应弛豫系统的总能量.根据表1中列出了所有Hf掺杂系统的位置能,Hf占据间隙位时位置能为-46.35 eV,Hf占据Zr位时的位置能为-39.12 eV和Hf占据Co位时的位置能为-40.40 eV.在Zr16Co16H48体系中,与Zr原子和Co原子位置相比,很显然Hf原子更喜欢占据Zr16Co16H48体系的间隙位,表明Hf原子较易进入Zr16Co16H48体内形成Zr16Co16HfH48化合物.
3.2 氢离解能
为了了解Hf对ZrCoH3脱氢的影响,氢离解能Ed由以下表达式计算[28],计算公式如式(2)所示:
Ed=[E(Zr16-xCo16-yHfx+y+zH47)+
(2)
式中x、y、z和E(Zr16-xCo16-yHfx+y+zH48)与(1)式定义相相同.在Zr16-xCo16-yHfx+y+zH47晶胞体系中包含一个氢空位,这是将Zr16-xCo16-yHfx+y+zH48晶胞体系中的一个氢原子移除而得到的.考虑到在Zr16-xCo16-yHfx+y+zH48晶胞体系中存在2类不同氢原子,如图1所示(H1、H2),将晶胞体系中的这2个氢原子分别从Zr16-xCo16-yHfx+y+zH48晶胞体系中移除,得到了含有两类氢原子空位的Zr16-xCo16-yHfx+y+zH47体系,由公式(2)可以计算出Zr16-xCo16-yHfx+y+zH48(M0、M1、M2和M3)体系的氢解离能,计算结果如表1所示,其中Ed-H1、H2分别表示形成H1、H2氢空位时的氢离解能.为了估算脱氢最大离解能,对Zr16-xCo16-yHfx+y+zH47晶胞体系不进行几何弛豫,即Zr16-xCo16-yHfx+y+zH47体系中的晶格参数和原子坐标保持与Zr16-xCo16-yHfx+y+zH48系统相同.相反,如果估算脱氢最低离解能,则对Zr16-xCo16-yHfx+y+zH47体系应该完全弛豫,表1中括号外数值代表脱氢最低氢离解能,括号里数值代表脱氢最大离解能.表1计算结果表明,Zr16-xCo16-yHfx+y+zH48(M0、M1、M2和M3)体系的氢解离能Ed-H1、-H2结果差异很明显,因此,Zr16-xCo16-yHfx+y+zH48晶胞体系的脱氢能力可以用Ed-H1、-H2的平均值Ea表征.对于Zr16-xCo16-yHfx+y+zH48体系,氢解离能Ed-H1、-H2存在差异,由此表明Zr16-xCo16-yHfx+y+zH48晶胞内存在两类不同的氢原子.如表1所示,与纯Zr16Co16H48脱氢能力相比,Hf掺杂带来了Zr16Co16H48氢离解能降低,尤其是当Hf原子占据Zr原子时.由此表明添加Hf原子在动力学上有利于Zr16Co16H48放氢,这与文献[13]的实验结果是一致的.将表1中的氢离解能与位置能进行比较,发现两种能量之间存在悖论,例如,具有-46.35eV最低占位能的间隙掺杂化合物 (Zr16Co16HfH48)显示出0.11 eV的最大氢离解能.虽然Hf原子掺杂时优先占据间隙位,但对ZrCoH3氢离解能的影响较小.相比之下,Hf原子很难占据Zr位置以产生Zr15Co16HfH48化合物,但这种化合物具有低氢离解能,相对容易放氢.
3.3 态密度
为了揭示Hf掺杂对Zr16-xCo16-yHfx+y+zH48体系放氢性能影响及体系组成原子Zr、Co、H 与 Hf之间的价键特征,图2分别给出了Zr16-xCo16-yHfx+y+zH48体系的总态密度图(DOS)、分波态密度图(PDOS),将图中费米能级Ef位置点作为能量零点.在图2中DOS图中出现了分别被标记为I、II、III 的3个主要的键峰.对于Zr16Co16H48(图2a),峰Ⅰ、峰Ⅱ主要由两类H原子的s态电子和Co的d态电子组成;费米能级以上的峰Ⅲ主要来自Zr的d电子和Co的d电子贡献组成.显然,Co和H原子轨道之间存在强烈的共价杂化,这与文献[19,23]结论是一致的,并且Co-H共价作用是Zr16Co16H48化合物结构稳定的主导因素.在费米能级处Zr的DOS值较小,Zr与H的之间的键合作用表现出弱的共价作用.在无Hf系统 (M0)中表现出的Co-H 共价性质和Zr-H 弱的共价性质也可以在Hf掺杂系统 (M1-M3)中观察到.Hf掺杂导致TDOS峰值降低,如由两类H原子的s态电子和Co的d态电子贡献的峰Ⅰ分别为:31.87856 electrons/eV(图2a M0)、30.09375 electrons/eV(图2b M1)、31.66705 electrons/eV(图2c M2)、31.75483 electrons/eV(图2d M3),由两类H原子的s态电子和Co的d态电子贡献的峰Ⅱ分别为:68.95441 electrons/eV(图2a M0)、65.61569 electrons/eV(图2b M1)、65.85231 electrons/eV(图2c M2)、67.63608 electrons/eV(图2d M3)和由Zr的d态电子和Co的d态电子贡献峰Ⅲ分别为:20.07668 electrons/eV(图2a M0)、18.67979 electrons/eV(图2b M1)、18.69867 electrons/eV(图2c M2)、18.8848 electrons/eV(图2d M3).这表明Co和H原子之间的共价s、d杂化被Hf掺杂削弱.本文中,Co-H共价杂化的削弱无疑有利于Zr16Co16H48的脱氢.因此,Hf掺杂削弱Zr16Co16H48的Co-H价键作用,从而降低Zr16Co16H48的结构稳定性,并改善Zr16Co16H48的放氢能力.从图2可以发现,费米能量附近的 Hf-s、-p、-d电子的键合作用峰倾向于与其相邻的Zr、Co和H 原子成键,因为该峰与基体中的Zr、Co和H 原子轨道重叠,因此Hf-H之间的存在键合作用.然而,在Hf取代体系中(M1、M2、M3),Hf与相邻Zr、Co原子的键长达到3.66-7.78 Å,3.86-6.78 Å,Hf-Zr、-Co之间的键合作用可以忽略不计.
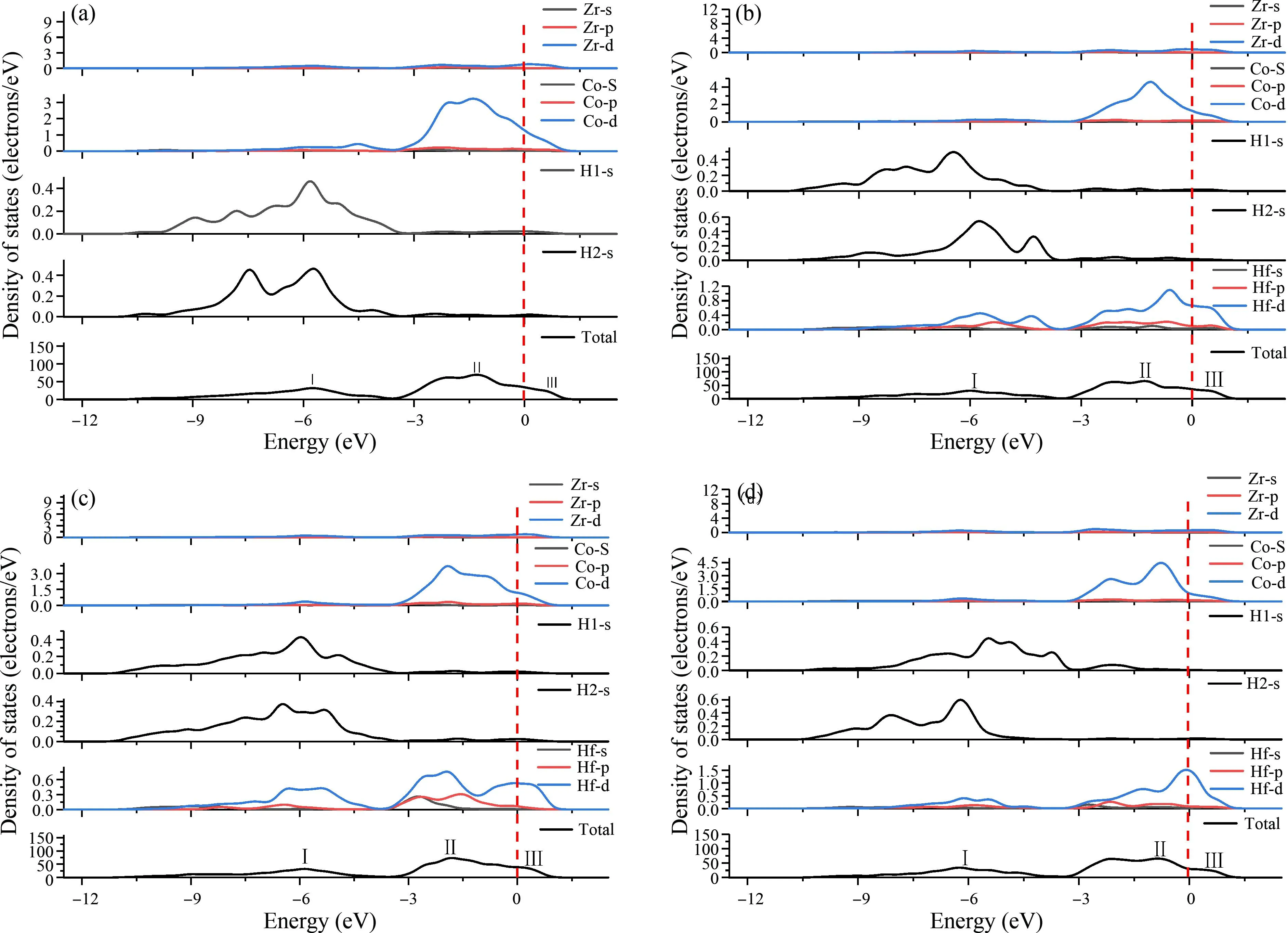
图2 Zr16-xCo16-yHfx+y+zH48体系总态密度(TDOS)以及组成原子分波态密度(PDOS)
3.4 电子密度
材料化学组成和微观结构的不同,决定其有不同的特性,材料在内部分子层次上如原子、电子之间的相互作用和化学成键对材料性能产生决定性的影响,电荷密度分布是研究成键特征的另一种直观方式.图3给出了Zr16-xCo16-yHfx+y+zH48晶胞体系在Zr、Co、H或/和Hf原子位点的电荷密度分布结果,图3中等高线处于0.003~0.6 electrons/Å3之间,彩色条形图显示了整个图中的相对电子密度.图3可见,在Zr16-xCo16-yHfx+y+zH48晶胞体系中Co原子周围的相对电荷密度高于Zr原子周围的相对电荷密度,这与图2是吻合的,Co、H、Zr原子的电子云重叠,表明Co-H、Zr-H、Zr-Co以共用电子的形式相互成键,显示出Co-H、Zr-H、Zr-Co的共价特征,并且由于Co-H周围电荷密度分布的方向性特征,Co和H原子之间存在强极化共价键.虽然Zr原子与H、Co原子之间有电子重叠,但电子云重叠密度远低于Co与H原子间相互电子云重叠密度,表明Zr-Co、Zr-H共价键强远低于Co-H共价键强,显示弱共价性.由于Hf占据Zr、Co位和间隙位时,Hf和H之间出现明显的重叠电荷,即形成Hf-H键.电子密度等值线中描述的Zr、Co、H和/或Hf原子之间的键合特性与DOS分析非常吻合.图3可以发现,随着Hf的添加,Co与H之间的电荷重叠在减弱,Co-H的键强在削弱,有利于Zr16-xCo16-yHfx+y+zH48放氢的发生.

图3 Zr16-xCo16-yHfx+y+zH48体系的电子密度图
3.5 Mulliken电荷布局
为了定量阐明成键特性,对Zr16-xCo16-yHfx+y+zH48体系晶胞进行了布居分析(Mulliken),包括平均键序 (BO),平均键长 (BL)和平均单位键长的键序 (SBO).SBO为单位键长的平均键序,即SBO= BO/BL,SBO能够用来比较两个原子的键强,如果SBO>0,则两个原子成键具有共价键性质,而且SBO值越高,成键相互作用越强[29,30].根据Mulliken电荷布局分析,表2列出了Zr、Co、H和Hf中主要的BO、BL和SBO值.从表2可以看出,Co-H、Zr-H、Zr-Co和Hf-H键的平均单位键长的键序为正值(SBOCo-H>0、SBOZr-H>0、SBOZr-Co>0和SBOHf-H>0),表明Co-H、Zr-H、Zr-Co和Hf-H键具有共价特征,该结果与图2、图3描述一致.由态密度和电子密度分析可知,Zr16Co16H48的稳定结构源于体内存在强烈的Co-H共价作用.Mulliken 布局分析表明,Hf掺杂后Hf原子占据掺杂位的不同,Co-H (SBOCo-H)之间的键阶从最大值0.14 Å-1降低到最小值0.11 Å-1,顺序为Zr16Co16H48> Zr16Co16HfH48> Zr16Co15HfH48> Zr15Co16HfH48,导致Zr16Co16H48结构的“失稳”,并降低Zr16Co16H48的氢离解能.这个降序恰好与表1中氢离解能Ed的降序一致.因此,Hf原子掺杂削弱了Co-H键强,并改善了ZrCoH3的脱氢动力学[13].此外,由于键序降低,键长增加,SBOZr-H和SBOZr-Co也因Hf掺杂而减小.然而,相对于Hf掺杂占据间隙位(SBOCo-H=0.13 Å-1、SBOZr-H=0.04 Å-1、SBOZr-Co=0.03 Å-1),Hf掺杂占据Co位(SBOCo-H=0.12 Å-1、SBOZr-H=0.03 Å-1、SBOZr-Co=0.0008 Å-1、SBOHf-H=-0.01 Å-1),Hf掺杂占据Zr位Co-H、Zr-H、Zr-Co、Hf-H作用较强,但脱氢能耗较低(SBOCo-H=0.11 Å-1、SBOZr-H=0.03 Å-1、SBOZr-Co=0.01 Å-1、SBOHf-H=-0.06 Å-1).由此表明,Co-H、Zr-H、Co-Zr键作用并非决定Hf掺杂的Zr16-xCo16-yHfx+y+zH48体系脱氢能力的最关键因素.本研究发现,掺杂的Hf原子与其周围的H原子发生相互作用生成Hf-H键,由此可知,Hf-H键作用对体系脱氢起到催化效果,而且这种催化对放氢起到主导作用,因为占据Zr位时Hf原子与其周围的H原子发生相互作用生成Hf-H键催化使具有较高Co-H、Zr-H、Zr-Co键作用,而表现出良好的放氢特性.

表2 Zr16-xCo16-yHfx+y+zH48和Zr16-xCo16-yHfx+y+zH47体系 Mulliken 布局分析得到平均键序,平均键长(单位Å)及平均单位键长的键序(单位Å-1)
3.6 能带结构
图4展示了Zr16-xCo16-yHfx+y+zH48晶胞的能带结构,其中费米能级设置为零能量,带隙(Eg)表征为导带最低能量和价带最高能量之间的间隙,如表1、图4所示Eg=0.显然,对于主体化合物Zr16-xCo16-yHfx+y+zH48价带和导带明显重叠,并且在费米能级处没有带隙,如图4所示,因此Zr16-xCo16-yHfx+y+zH48具有金属性质,这与文献[14]报道是一致的.因此,Zr16Co16H48及Hf掺杂后的Zr16-xCo16-yHfx+y+zH48化合物倾向于表现出金属的特性,从而导致分解Zr16-xCo16-yHfx+y+zH48的能量成本较低,因为金属系统被认为是有利于改善Zr16-xCo16-yHfx+y+zH48的脱氢动力学[31].为了了解Hf-H键对Zr16-xCo16-yHfx+y+zH48体系脱氢的影响,我们在M1、M2和M3体系中引入了一个最佳H原子空位,为脱氢的化学反应创造有利条件,因为传质化学反应进行仅通过晶格缺陷发生,并且发现氢扩散是由氢空位介导[32].在此,认为与最低脱氢能相对应为最佳H原子空位,在母系统中很容易诱导,即M1系统的H2空位和M2、M3系统的H1空位.由表2的计算结果发现对于所有Hf掺杂体系,Co-H、Zr-H和Hf-H之间的键合数在引入H空位前后略有变化,对于M1、M2系统由于H原子被带走,Hf和H之间的相互作用从弱离子性质转移到强共价性质,由于BOHf-H从-0.23、-0.04增强到0.25、0.15,BLHf-H从3.83Å、2.77Å增加到2.21Å、2.91Å,进而SBOHf-Co从-0.06 Å-1、-0.01 Å-1增强到0.11 Å-1、0.05 Å-1.因此在M1和M2体系中,Hf和H之间的相互作用可以提高Zr16-xCo16-yHfx+y+zH48体系的放氢性能.对于M3体系,H空位的引入Hf-H键缺失不利于M3化合物放氢.
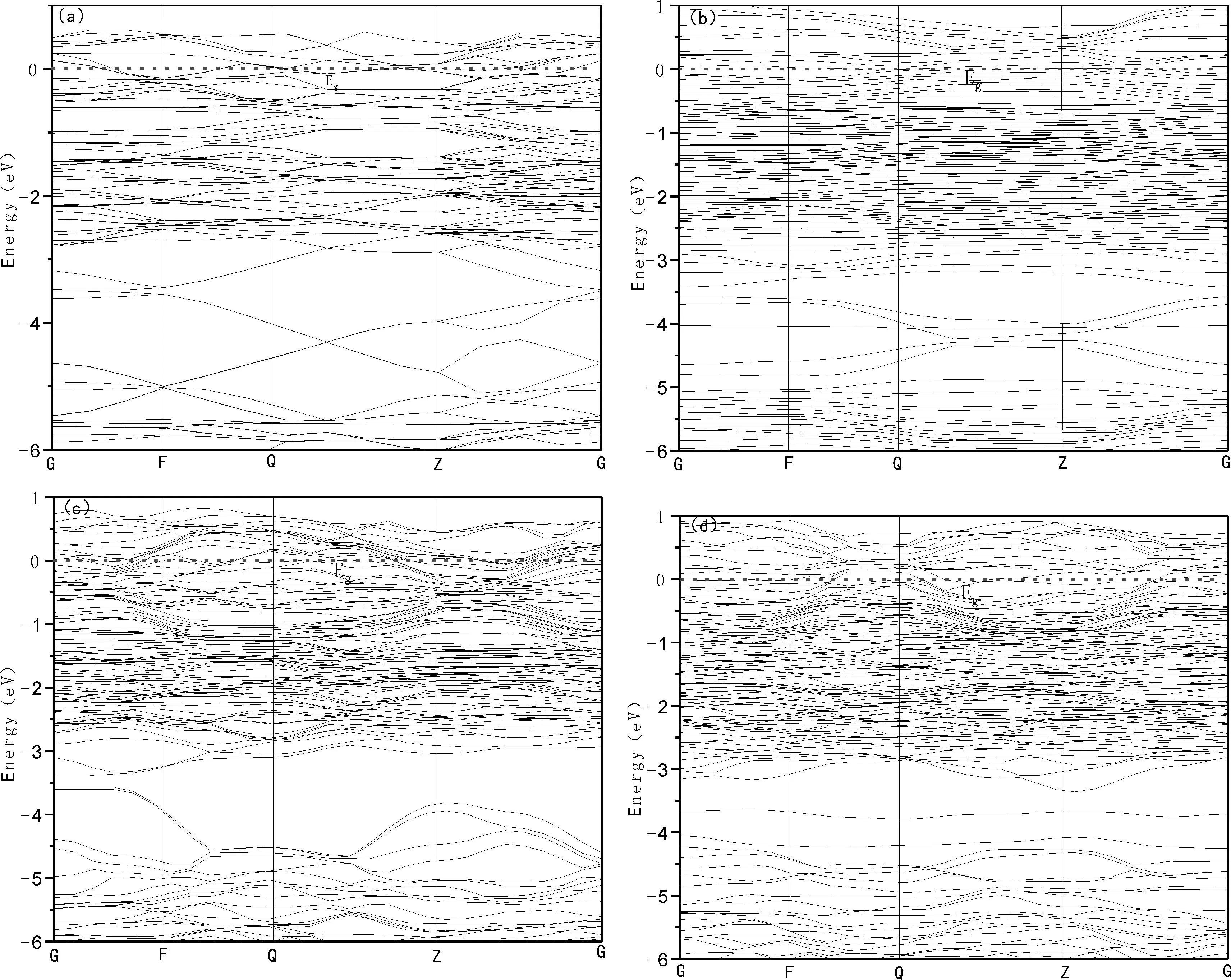
图4 Zr16-xCo16-yHfx+y+zH48能带结构
4 结 论
在本研究中Hf原子被设计为通过占据Zr原子、Co原子和间隙位点来添加到Zr16Co16H48中.通过第一性原理密度泛函理论计算研究了Hf原子掺杂的选择能、氢离解能、电子结构以及无Hf和Hf掺杂的Zr16Co16H48的键合特性,旨在研究Zr16Co16H48-Hf体系的放氢行为,计算结果表明:
(1)Hf掺杂弱化了Co-H、Hf-H的共价键强度,降低了Zr16Co16H48的结构稳定性,键强有利于提高Zr16Co16H48系统的放氢能力,导致氢离解能(Ed)、Co-H平均单位键长的键序(SBOCo-H)均按Zr16Co16H48> Zr16Co16HfH48> Zr16Co15HfH48> Zr15Co16HfH48次序降低.
(2)Hf掺杂优先占据间隙位产生Zr16Co16HfH48化合物,但是这种具有较高氢离解能的化合物与其他两种Hf掺杂化合物相比结构比较稳定.虽然Zr15Co16HfH48表现出良好的放氢性能,但实际应用中应降低Hf占据Zr位的能量成本.
(3)通过本研究发现Zr16Co16H48系统的脱氢能与位置能,这两种能量之间存在悖论关系.
致 谢:作者感谢四川大学建筑与环境学院曾祥国教授提供的计算资源.
猜你喜欢
高中数理化(2022年16期)2022-09-14
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
高中数理化(2022年4期)2022-03-14
潍坊学院学报(2021年2期)2021-07-22
高等学校化学学报(2021年7期)2021-07-11
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
山东化工(2019年1期)2019-01-24
东华大学学报(自然科学版)(2018年1期)2018-06-29
信息记录材料(2016年4期)2016-03-11
