
UPLC-MS法测定肉制品中虾过敏原含量的不确定度评定
2024-01-25 12:07梁瑞强刘彤彤罗娇依孙姗姗
生物加工过程 2024年1期
梁瑞强,刘彤彤,罗娇依,曹 进,孙姗姗
(中国食品药品检定研究院 国家市场监管重点实验室(食品质量与安全),北京 100050)
蛋白类抗原是引起食物过敏的主要因素,其中虾肉又属于主要的过敏食物[1-2]。在临床上,曾有因食用虾而导致过敏的报道,虾肉过敏可引起皮肤刺激和消化系统紊乱,更严重的情况会导致血管性水肿和休克[3]。虾过敏原主要包括原肌球蛋白、肌质钙结合蛋白和血蓝蛋白等,因此,针对这些过敏原的性质特点,免疫学技术、色谱质谱联用技术和分子生物学技术等分析虾过敏原的方法相继出现[4-8]。目前,我国的相关标准要求:对可能引起过敏反应的食品及其制品进行相关标注和提示[9]。
对于食品过敏原的检测,技术人员常采用实时链式聚合酶反应法(RT-PCR)或酶联免疫吸附法(ELISA)[10]。RT-PCR是通过测定与过敏原蛋白质相关的DNA片段而得到过敏原蛋白的含量,若过敏原蛋白的基因序列中有未知基因序列,则该方法不适用。ELISA法的基本原理是抗体和抗原的相互作用,如果样品中非过敏原蛋白与抗体产生交叉反应,则会影响检测结果的准确性。此外,光谱法、电泳法、比色法和液相色谱法等虽然都可用于蛋白质含量测定,从而应用于食品过敏原检测,但对于基质复杂的样品也不适用。相比于上述技术手段,超高效液相色谱串联质谱法(UPLC-MS)不仅可以通过离子质荷比(m/z)的差异同时鉴定多种蛋白质,还能通过测定目标肽段来定量蛋白质含量,具有灵敏度高、专属性强、容易实现自动化进样的优势[11]。采用UPLC-MS法进行过敏原定量分析不仅可以避免ELISA法检测时可能出现交叉反应的缺点,还能一定程度上弥补RT-PCR法遇到未知基因序列而无法定量的不足。通常,使用质谱技术研究食品过敏原的对象主要集中在含有牛奶、坚果类和大豆成分的食品。如,含有牛奶的饮料、水果制品和烘烤类食品,含有各类坚果(核桃仁、杏仁等)的预包装食品,含有大豆成分的复杂加工食品等,但对于肉制品中虾类过敏原的研究相对较少。因此,基于食品安全监管需求、技术优势和现有研究的短板,采用UPLC-MS技术开发肉制品中虾过敏原的检测方法是一个具有前景的方向。
由于蛋白质具有分子量大、电荷多等特点,单纯依靠质荷比(m/z)并不能直接实现对蛋白质的鉴定,需要采用合适的酶解方法将目标蛋白质降解成肽段,再从中寻找能够代表蛋白质结构和含量特点的专属性肽段(特征肽段)进行分析。笔者课题组刘彤彤等[12]前期研究利用蛋白质组学相关技术获得肉制品中虾过敏原的特征肽段,并完成了使用UPLC-MS技术检测过敏原含量方法的开发。对于具体含量测定方法,不仅需要保证使用此方法进行检测所得结果的准确度和可信程度,而且还要评定测量不确定度,以使测定方法的结果较为客观。
因此,本研究的目的在于通过测量不确定度来表征所开发的UPLC-MS检测结果的合理离散度并评估测量结果的质量,并建立该方法测定肉制品中虾过敏原含量的不确定度数学模型,分析实验过程中引入不确定度的分量和占比,得到对检测结果产生影响的主要因素,以期为在实际应用场景中使用此方法的操作过程提供合理建议,也为提高实验结果的准确度和置信度提供依据。
1 材料与方法
1.1 仪器
UPLC-Xevo TQ-S型超高效液相色谱串联三重四极杆质谱仪、胰蛋白酶(质谱级,1 mg),美国沃特世公司;Synergy HT型酶标仪,德国BioTek 公司;B-491型恒温水浴锅,瑞士步琪公司;AL204、XP205型电子天平,瑞士梅特勒-托利多公司;SORVALL Legend MICRO21 R型离心机,美国赛默飞世尔科技公司;ZWY-240型涡旋振荡器,美国Scientific Industries 公司。
1.2 试剂
甲酸、乙腈(质谱纯)、胰蛋白酶(质谱级,1 mg),美国赛默飞世尔科技公司;三羟甲基氨基甲烷(分析纯),美国西格玛奥德里奇公司;磷酸盐缓冲液(PBS)片剂,北京索莱宝科技有限公司;盐酸(分析纯),国药集团化学试剂有限公司;Bradford 蛋白测定试剂盒,美国伯乐公司;对照品肽段:IR-9(序列为IVELEEER,纯度≥97.05%,批号为P200720-XT642878),内标肽段为IR-9*(序列为IVEL*EEER,纯度≥95.40%,批号为P200720-XT818725),上海吉尔生化有限公司;实验用水由H2O pro-DI-B型超纯水器(德国赛多利斯公司)制备。
PBS溶液:取PBS 片剂1片加 100 mL 超纯水超声溶解后,混匀即得。
胰蛋白酶溶液:用 1 mL 超纯水溶解胰蛋白酶即得,质量浓度为 1 mg/mL。
1.3 样品
从京东商城购买包含虾类成分的肉制品(包括不同品牌的香肠、肉丸和火腿等)作为样品,共20批;空白样品(牛肉、羊肉、猪肉、鸡肉、鸭肉、鲈鱼、黑鱼、鲤鱼、黄鳝等比例混合)购于北京物美超市。由于虾类属于节肢动物门生物,与脊索动物门中哺乳纲、鸟纲和硬骨鱼纲的亲源性较远,故选为阴性基质;另因以上肉类常作为混杂肉类制成熟食预包装食品,故选为研究基质对象。所有样品储存于-20 ℃冰箱。
1.4 仪器及分析条件
在前期的方法开发工作中,通过蛋白质组学技术得到了虾过敏原蛋白的特征肽段,并合成了对应的同位素内标肽段。将特征肽段作为对照品(外标),同位素内标肽段作为内标,使用UPLC-Xevo TQ-S质谱自带的IntelliStart技术自动得到内标和外标的质谱条件和多反应监测(MRM)通道参数,并根据在超高效液相色谱上外标、内标的色谱行为优化得到合适的色谱条件。
具体色谱条件为色谱柱UPLC HSS T3(150 mm×2.1 mm,1.8 μm,美国沃特世公司)、柱温30 ℃、进样量5 μL、流速0.3 mL/min。流动相:A相为0.1%甲酸-水溶液(体积分数),B相为乙腈。梯度洗脱程序:0 min,V(A)∶V(B)=82∶18;0.5 min,V(A)∶V(B)=82∶18;8.0 min,V(A)∶V(B)=55∶45,8.3 min,V(A)∶V(B)=18∶82;8.6 min,V(A)∶V(B)=18∶82 ;9.0 min,V(A)∶V(B)=82∶18;11.5 min,V(A)∶V(B)=82∶18。
质谱条件为利用正离子扫描方式的电喷离子源(ESI+),扫描模式为多反应监测(MRM),毛细管电压3.2 kV,去溶剂气温度500 ℃,离子源温度150 ℃,脱溶剂气流量800 L/h,锥孔气流量为150 L/h。MRM通道参数见表1,其中565.5→917.6为外标肽段IR-9的定量离子对,569.0→924.7为内标肽段IR-9*的定量离子对。
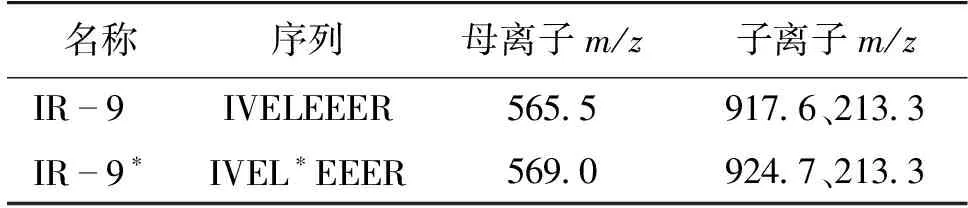
表1 虾过敏原肽段MRM通道参数
1.5 对照品溶液的制备
在前期方法开发阶段,根据样品中待测成分的大致含量范围确定了标准工作溶液的工作范围,从而得到对照品溶液的制备方法。
分别精确称取1.04 mg外标IR-9、0.76 mg内标IR-9*,用1 mL Tris-HCl(50 mmol/L)溶液溶解,得到外标溶液IR-9浓度为893.8 μmol/L、内标溶液IR-9*浓度为638.1 μmol/L。再分别吸取外标溶液112.0 μL,用50 mmol/L Tris-HCl溶液定容至5 mL,内标溶液31.3 μL,用50 mmol/L Tris-HCl溶液定容至2 mL,得到外标储备溶液浓度为20 μmol/L、内标储备溶液浓度为10 μmol/L。进一步稀释得到浓度为5 μmol/L外标中间溶液和浓度为2.5 μmol/L的内标中间溶液。取适量外标中间溶液,再加入内标中间溶液,用水稀释定容,分别配制成外标浓度为0.001、0.01、0.1、0.5和2.0 μmol/L的标准工作溶液(各浓度工作溶液均含有0.1 μmol/L内标溶液)。
1.6 样品溶液的制备
根据样品的性质特点并参照文献[11]所述样品处理方法,对样品前处理条件进行研究,得到了本研究的样品溶液制备方法。
在50 mL离心管中精密称取样品5 g。用移液器准确加入30 mL PBS溶液,超声提取30 min。设置10 000 r/min的转速,温度为15 ℃,离心10 min。量取上清液5 mL,加乙腈5 mL,涡旋振荡1 min后静置,弃上清液,用1 mL PBS溶液复溶残渣,并经0.45 μm滤膜过滤。准确移取上述滤液500 μL,加入40 μL 2.5 μmol/L内标溶液、10 μL胰蛋白酶液,涡旋混匀,于37 ℃恒温水浴中反应16 h,取出并加入体积分数10%的甲酸-水溶液10 μL以终止反应,补超纯水至1 mL。设置10 000 r/min的转速,温度为4 ℃,离心10 min,得到待分析的上清溶液。
1.7 数学模型的建立
据《测量不确定度表示指南》规定:测量不确定度评定步骤时,首先要建立数学模型,具体数学模型应当包含所有对测量结果的不确定度有影响的修正值和修正因子。在测量中,当被测量值(即输出量)由其他量(即输入量)通过函数来确定时,可以得到一个数学模型,这样建立的数学模型可用来计算测量结果,也能用来评定测量结果的不确定度。实际情况中,对于一些检测方法,虽然有些输入量对测量结果有影响,但由于信息量的缺乏,在具体测量时无法定量计算它们对测量结果的影响,从而无法得到与输出量相关的解析表达式;也有些输入量由于对测量结果的影响很小,从而可以被忽略不计,这时测量不确定度评定的数学模型才可以与计算公式相同。
根据样品处理过程,得到虾过敏原含量的计算公式式(1),此式描述了被测量(虾过敏原含量,即输出量)与酶解液中外标浓度(输入量)之间的函数关系,因此将式(1)作为本次不确定度的评定数学模型。
(1)
式中:X为每1 kg样品中虾过敏原的质量,mg/kg;c为酶解溶液中外标的浓度,μmol/L;V1为提取样品时加入PBS溶液的体积,mL;V2为酶解溶液的定容体积,mL;V3为待酶解样品提取溶液的体积,mL;m为称样量,g;M为虾过敏原(蛋白质)的摩尔质量,32 663 g/mol;1 000为换算系数。
1.8 不确定度来源分析
综上可知,本研究方法的不确定度来源主要包括样品称量、重复性试验、方法准确度、样品前处理过程和样品溶液测定过程,同时每个过程引入的不确定度又有各自的分量,结合文献[13-14]将这些不确定度来源和相互关系用“鱼骨图”表示,具体见图1。
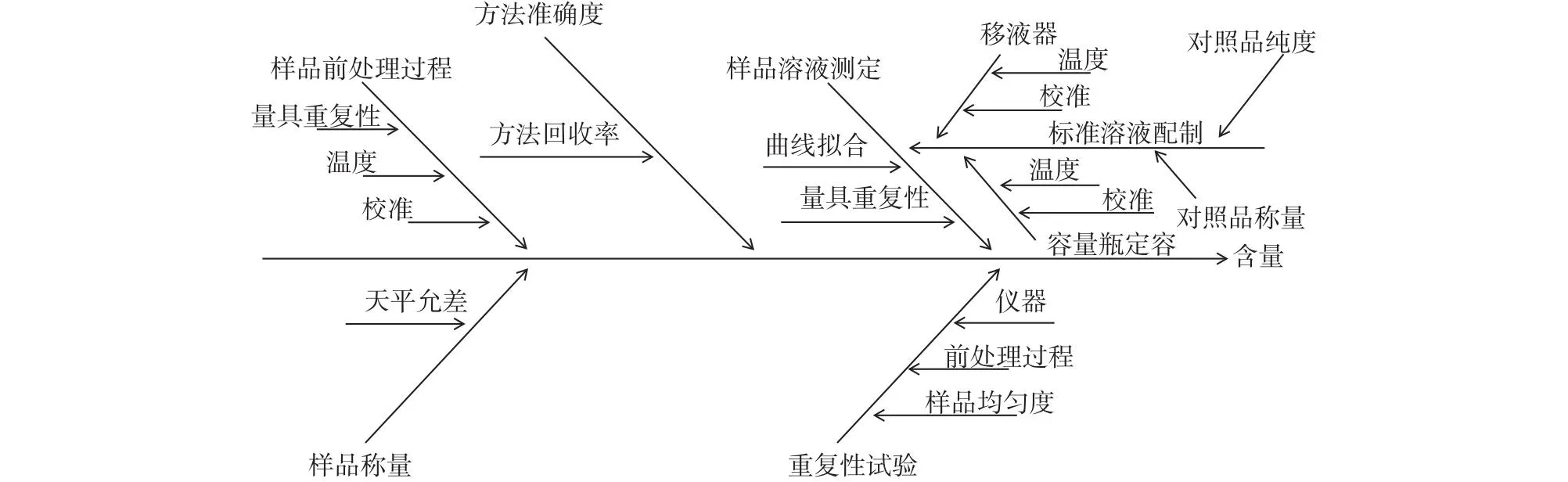
图1 不确定度来源分析Fig.1 Analysis of uncertainty sources
1.9 样品称量引入的合成相对标准不确定度um,rel
本研究用使用万分之一电子天平称样,每次称量会引入相对标准不确定度,而样品称量引入的相对标准不确定度由多次称量的不确定度合成。单次称量的相对标准不确定度以及样品称量引入的合成相对标准曲线不确定度的计算见式(2)~(3)。
(2)
式中:um,rel(n)为单次称量的相对标准不确定度;um为单次称量的标准不确定度,g。
(3)
式中:um,rel为供试品称量过程引入的合成相对标准不确定度。
1.10 重复性试验引入的相对标准不确定度ur,rel
平行称量同一肉制品样品6份,按1.6所示方法处理,得到6次测定结果的均值。由于此不确定度分量对应的测量值属于多次独立重复测量的结果,可按照A类不确定度评定[15],具体计算见式(4)。
(4)
式中:ur为重复性试验引入的标准不确定度,mg/kg;ur,rel为重复性试验引入的相对标准不确定度;Sr为n份平行样品含量测定结果的标准偏差,mg/kg;ci为单份样品的含量测定结果,mg/kg;c0为n份平行样品含量测定结果的平均值,mg/kg;n为样品重复性试验测定的次数。
1.11 方法准确度引入的相对标准不确定度ua,rel
方法准确度一般用回收率来表示。本次回收率试验过程中选取3个浓度水平进行加标,在每个浓度水平中平行处理6份样品,共得出回收率试验数据18个,可计算得到回收率的平均值。按照显著性检验(t检验),若回收率的平均值与100%的差异有统计学意义,引入的不确定度即用回收率平均值的标准偏差计算[13],具体计算见式(5)。
(5)
式中:ua为回收率平均值的标准不确定度,%;ua,rel为方法准确度引入的相对标准不确定度;Sa为回收率平均值的标准偏差;ai为单次回收率,%;a0为回收率的平均值,%;n为回收率测定的次数(n=18)。
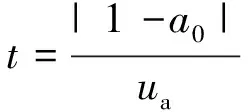
1.12 样品前处理过程引入的相对标准不确定度uxv,rel
样品的前处理过程用到了不同量程的移液器进行吸液,分别为1~10 mL移液器吸液10 mL,0.5~5 mL移液器吸液5 mL,100~1 000 μL移液器分别吸液1、0.5和0.44 mL,10~100 μL移液器吸液40 μL(0.04 mL)以及0.5~10 μL移液器吸液10 μL(0.01 mL),共经7次操作,具体计算见式(6)。
(6)
式中:uxv,rel为前处理过程取引入的相对标准不确定度;u(v1),u(v2),…,u(vi)分别为用移液器吸液体积为v1,v2,…,vi时的引入的相对标准不确定度;n1,n2,…,ni为用移液枪进行吸液v1,v2,…,vi的操作次数。而每次吸液操作引入的相对标准不确定度又由移液器的校准u1(vi)、温度效应u2(T,vi)和吸液体积重复性u3(vi)引入的相对标准不确定度3个分量合成得到。
u1(vi)的计算见式(7)。
(7)
式中:u1(vi)为移液器校准引入的相对标准不确定度;Evi为移液器对应吸液体积的允许误差,mL;vi为移液器对应的吸液体积,mL;k为包含因子。
u2(T,vi)的计算见式(8)。
(8)
式中:V为量具取液或者定容的体积,mL;ΔT为实验环境的实际温度与实验室标准温度(20 ℃)的偏差,℃(本次取值为5 ℃);2.1×10-4为水的膨胀系数,℃-1。
u3(vi)的计算见式(9)。
u3(vi)=Svi/vi
(9)
式中:Svi为用移液器吸取样品前处理操作中对应的各吸液体积的水10次并进行称量的结果标准偏差,mL;vi为用移液器吸取样品前处理操作中对应的各吸液体积,mL。
1.13 样品溶液测定过程引入的相对标准不确定度uxc,rel
样品溶液测定过程引入的相对标准不确定度由对照品纯度up,rel、对照品称量uω,rel、标准溶液配制us,rel和标准曲线拟合uL,rel这4部分相对标准不确定度分量合成得到,具体计算见式(10)。
(10)
up,rel的计算见式(11)。
(11)
式中:up,rel为对照品纯度引入的相对标准不确定度;Ep为对照品纯度允许误差,%;p为对照品纯度,%。
uω,rel的计算见式(12)。
(12)
式中:uω,rel为对照品称量引入的相对标准不确定度;uω为对照品称量的标准不确定度;ω为称取对照品的质量,g。
本实验在标准溶液配制过程中进行了100~1 000 μL移液器吸液1 mL、0.5 mL,10~100 μL移液枪吸液0.1 mL,20~200 μL移液枪吸液185.8 μL、112.0 μL,0.5~5 mL移液枪吸液2.5mL,10 mL容量瓶定容,5 mL容量瓶定容,共8种操作。因此us,rel的计算见式(13)。
(13)
式中:us,rel为标准溶液配制过程中产生的相对标准不确定度;uv1、uv2、…、uvi分别为使用不同量具(不同规格移液器或容量瓶)的吸液或定容体积为v1、v2、…、vi时的产生的相对标准不确定度;n1、n2、…、ni为吸液或定容体积为v1、v2、…、vi时对应量具的使用次数。每次吸液或定容的操作产生的相对标准不确定度又由量具的校准u1(vi)、温度效应u2(T,vi)和量具体积重复性u3(vi)的相对标准不确定度合成得到。对应的计算方法参照1.12中的公式(7)~(9)。
uL,rel的计算见式(14)~(16)。
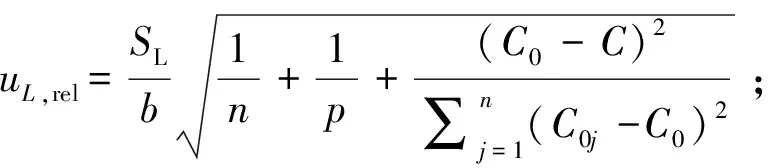
(14)
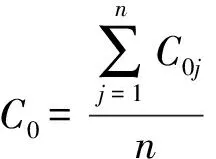
(15)
(16)
式中:SL为标准曲线的剩余标准差;a和b分别为标准曲线的截距和斜率;n为拟合标准曲线的浓度点数;p为样品的测定次数;C0为标准曲线各点浓度的平均值;C为样品溶液中待测成分浓度的平均值;C0j为标准曲线各点的浓度;Aaj为各标准曲线各点的实际响应值;Aj为根据回归方程计算的各点理论响应值;uL为标准曲线拟合的标准不确定度;uL,rel为标准曲线拟合过程引入的相对标准不确定度。
1.14 方法的合成相对标准不确定度和扩展不确定度
方法的合成相对标准不确定度(uc,rel)由各相对标准不确定度分量通过式(17)计算。
(17)
方法的相对扩展不确定度(Urel)计算见式(18)。
Urel=kuc,rel
(18)
式中:uc,rel为方法的合成相对标准不确定度。
方法的合成标准不确定度(uc)的计算见式(19)。
uc=uc,relX
(19)
式中:X为重复性试验测定结果均值。
方法的扩展不确定度(U)计算见式(20)。
U=kuc
(20)
2 结果与讨论
2.1 um,rel计算结果
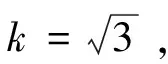
2.2 ur,rel计算结果
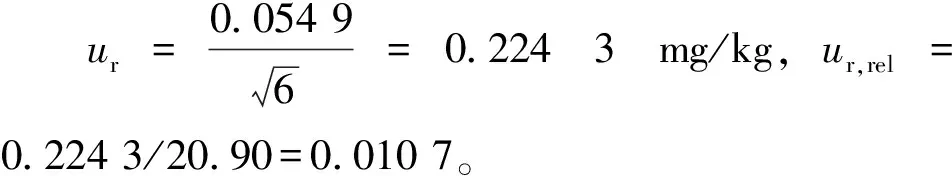
2.3 ua,rel计算结果
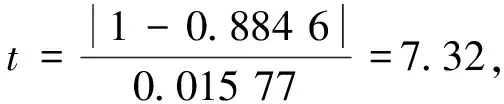
2.4 uxv,rel计算结果
2.4.1 移液器的校准引入的相对标准不确定度u1(vi)
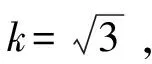
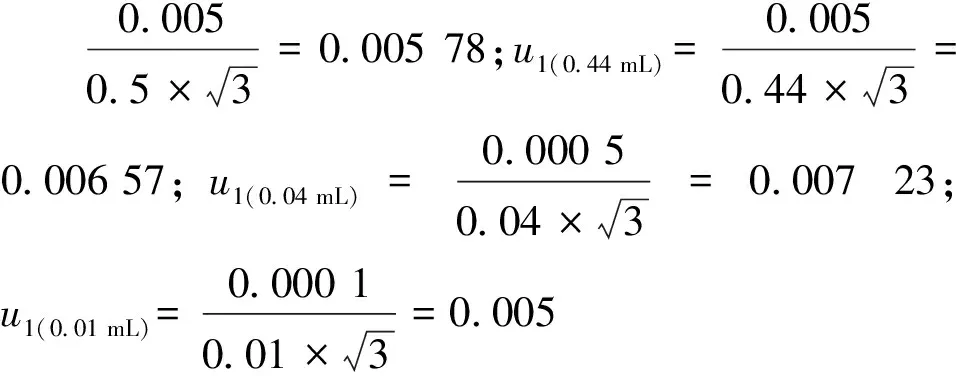
2.4.2 温度效应引入的相对标准不确定度u2(T,vi)
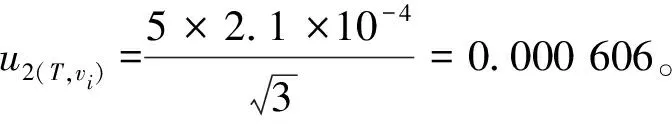
2.4.3 吸液体积重复性引入的相对标准不确定度u3(vi)
据式(9)计算,u3(10 mL)=0.007 59,u3 (5 mL)=0.000 314,u3(1 mL)=0.000 895,u3(0.5 mL)=0.001 21,u3(0.44 mL)=0.001 06,u3(0.04 mL)=0.001 43,u1(0.01 mL)=0.007 67。
2.4.4 前处理过程引入的相对标准不确定合成
根据2.4.1~2.4.3节的计算结果,可以合成不同移液器在各自吸液体积时的相对标准不确定度,具体计算如下
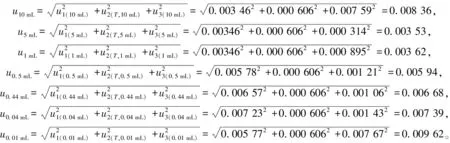
结合前处理过程中不同移液器吸液不同体积操作的次数,据式(6)合成前处理过程中引入的相对标准不确定度,具体结果为
2.5 uxc,rel计算结果
2.5.1 对照品纯度和对照品称量引入的相对标准不确定度up,rel、uω,rel
2.5.2 标准溶液配制引入的相对标准不确定度us,rel
对于100~1 000 μL移液器,吸液1 mL、0.5 mL产生的相对标准不确定度在2.4.1节中已说明,分别为u1 mL=0.003 62,u0.5 mL=0.005 94。
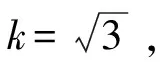
按照2.4.2所述,据式(8),温度效应引入的相对标准不确定度均为0.000 606。
体积重复性引入的相对标准不确定度按2.4.3节所述参照公式(9)计算的得到。各移液枪体积重复性不确定分别为u3(0.1 mL)=0.004 28,u3(185.8 μL)=0.003 05,u3(112 μL)=0.002 57,u3(2.5 mL)=0.000 903。容量瓶体积重复性不确定度计算过程为:分别对5 mL、10 mL容量瓶用超纯水定容,称质量,重复此操作10次,用得到的称量数据计算标准偏差,进而得到相对标准不确定度:u3(5 mL A)=0.000 333;u3(10 mL A)=0.001 50。
对每次吸液或定容的操作产生的相对标准不确定度的合成,结果如下

根据式(13)合成标准溶液配制过程中引入的相对标准不确定度us,rel
2.5.3 标准曲线拟合引入的相对标准不确定度uL,rel
外标IR-9的标准曲线方程y=1.848 4x-0.008 6,相关系数r2=1(n=5,相关系数大于 0.999)。横坐标为外标的物质的量浓度,纵坐标为外标峰面积与同位素内标峰面积的比值乘以内标浓度。据式(14)~(16),标准曲线各点浓度C0j分别为0.001、0.01、0.1、0.5和2.0 μmol/L;Aaj分别为0.002 0、0.017 0、0.171 0、0.901 0和3.691 0;Aj分别为-0.006 8、0.009 9、0.176 2、0.915 6和3.688 2。重复性试验中样品溶液中待测成分浓度的平均值C为0.107 μmol/L,C0为0.522 2 μmol/L,b和a分别为1.848 4和-0.008 6。通过以上数据可计算得到uL=0.003 95 μmol/L。
2.5.4 样品溶液测定过程引入的相对标准不确定度合成
据式(10),结合2.5.1~2.5.3节的计算结果,对样品溶液测定过程引入的相对标准不确定度进行合成,uxc,rel的具体结果为
2.6 合成相对标准不确定度和扩展不确定度计算
据式(17)计算,由各不确定度分量合成相对标准不确定度uc,rel为
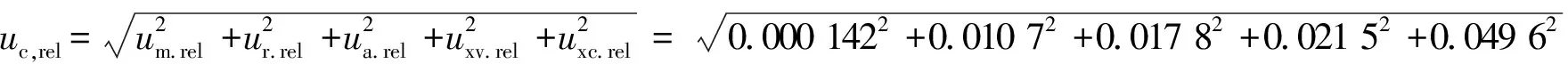
据式(18),取置信度为95%时的扩展因子k=2,得到方法的相对扩展不确定度为Urel=2×0.057 9=0.116。
据式(19),其中X取重复性试验测定结果均值,20.90 mg/kg,得方法的合成标准不确定度uc=0.057 9×20.90=1.21 mg/kg。
据式(20),仍取置信度为95%时的包含因子k=2,计算得到方法的扩展不确定度U=2×1.21=2.42 mg/kg。
因此,运用本方法定量分析肉制品中虾过敏原含量的结果为(20.90±2.42) mg/kg。
2.7 各不确定度分量的占比
综上可知,本方法引入的相对标准不确定度由样品称量、重复性试验、方法准确度、样品前处理过程和样品溶液测定过程5部分产生的相对标准不确定度分量合成得到,各不确定度分量的占比见表2。由表2可直观看出,用此方法进行定量分析,检测结果的不确定度主要来源于样品溶液测定过程。进一步观察全部不确定度评定过程可知,样品溶液测定过程不确定度的主要来源为对照品纯度、对照品称量、标准溶液配制和标准曲线拟合,占比分别为0.4%、33.5%、21.8%和44.4%。
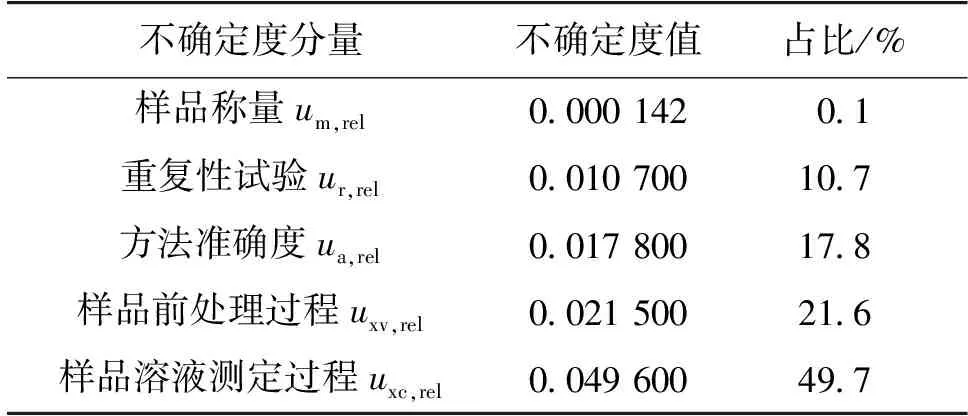
表2 UPLC-MS测定肉制品中虾过敏原含量的不确定度分量占比
2.8 分析讨论
结合不确定度评定结果和2.7节所示不确定度分量占比分析可知,对照品的称量和标准曲线拟合过程产生了较大的不确定度。
对于对照品的称量,本次实验采用的是十万分之一天平,称取1.04 mg,而十万分之一天平的实际分度值为0.01 mg,如果按照《中华人民共和国药典》(2020年版)凡例中的要求“‘精密称定’系指称取重量应准确至所取重量的千分之一”(注:此处的“重量”即为“质量”),则用该天平称样至少应为10 mg,才能使得称样品的千分之一不小于天平的实际分度值[20]。由于本次研究过程得到的合成肽段较少,可能导致不确定度的放大。因此,在实际检测过程中,称量对照品的质量应确保大于所使用天平的最小称样量。
对于标准曲线拟合过程,本实验标准工作溶液浓度为0.001~2.000 μmol/L,跨越3个数量级。这可能导致标准曲线各点浓度的平均值偏差的平方和变大,由式(14)可知,该值放大会增加标准曲线拟合的标准不确定度。因此,在实验过程中应根据样品中待测成分的实际含量合理配制标准曲线工作溶液的浓度范围。
从前期方法开发阶段对实际样品的检测结果可知,虾过敏原含量最低值2.3 mg/kg,最高值为448.1 mg/kg。依据GB/T 27404—2008《实验室质量控制规范 食品理化检测》可知,被测组分含量在1~1 000 mg/kg时,实验室内变异系数应在3.8%~11%(重复测定次数至少为6)[21]。在2.6节中所列方法的相对扩展不确定度为0.116(即11.6%),虽然实际检测时通常制备2份平行样,但为尽可能避免不确定度影响结果的准确性,所以在使用本方法检测样品中虾过敏原含量时,报告的检测结果最好带有与结果单位相同的测量不确定度[22]。
3 结论
本研究评定了用UPLC-MS法测定肉制品中虾过敏原含量过程存在的不确定度,发现定量分析结果的不确定度主要来源是样品溶液测定过程,而其中对照品的称量和标准曲线的拟合产生的不确定度较大,应在实验中采取增大对照品称样量和合理化标准曲线工作溶液浓度范围等方式以减少不确定度,并在报告检测结果时附带与结果单位相同的不确定度。本研究将所开发的虾过敏原含量测定方法中可能产生的不确定影响进行定量表示,可为后期准确测定肉制品中虾源过敏原的含量提供科学依据。
猜你喜欢
中国乡村医药(2023年16期)2023-09-08
河南科技(2023年1期)2023-02-11
Chinese Physics B(2022年5期)2022-05-16
中国动物保健(2022年2期)2022-05-05
技术与市场(2020年5期)2020-03-02
黑龙江交通科技(2020年5期)2020-01-13
有机氟工业(2019年2期)2019-08-12
实验与分析(2018年3期)2019-01-04
数字海洋与水下攻防(2018年1期)2018-10-29
实验与分析(2018年4期)2018-02-27
