
PdCo/准NH2-Ce-BDC串联催化氨硼烷脱氢和苯乙炔加氢
2024-02-04 09:47胡善红范建一邵帅李春生陈国柱
山东化工 2024年1期
胡善红,范建一,邵帅,2*,李春生*,陈国柱
(1.济南大学 化学化工学院,山东 济南 250024;2.山东鲁泰控股集团有限公司 石墨烯高分子复合材料研发中心,山东 济宁 272000)
苯乙烯是生产丙烯腈-丁二烯-苯乙烯共聚物、聚苯乙烯和丁苯橡胶的重要单体,但苯乙烯中苯乙炔的存在,不仅影响聚合过程中催化剂消耗量,而且影响聚合产物链长和聚合速率[1-2]。因此,除去苯乙烯单体中的苯乙炔对于制备高纯苯乙烯具有重要的工业意义。在高纯苯乙烯单体制备方法中,苯乙炔半加氢是获得苯乙烯最有效的途径之一。目前,Pd/C是催化苯乙炔半加氢反应使用最广泛的催化剂[3-4]。然而,单金属Pd活性位对氢有强亲和力,导致苯乙炔完全加氢生产副产物烷烃,降低了苯乙烯的选择性[5]。因此,亟需开发一种高活性、高选择性的苯乙炔半加氢催化剂。
研究表明,相比单金属催化剂,Pd-M(M为非贵金属)双金属之间的几何效应与电子效应能有效提升催化剂活性,同时贵金属合金化能有效降低催化剂生产成本,因而具有重要的工业意义。Pd-M双金属催化剂中,Pd-Co、Pd-Ni等在液相选择性加氢反应表现出高活性和高选择性[6-7]。除活性组分外,载体的选择同样是影响催化剂活性的关键因素之一。近年来,金属-有机框架材料(Metal-Organic-Frameworks,MOFs)因其丰富的不饱和金属位点、较好的热稳定性、丰富的孔结构,被广泛用作催化剂载体[8]。相比MOFs,通过部分去除有机配体获得的准MOFs不仅仍保留原有MOFs的结构特征,保留高比表面积和孔道结构,更重要的是,残留的有机配体影响反应底物的吸附,使得准MOFs成为一类独特的金属粒子载体。譬如,本课题组前期工作中利用准Ce-MOFs载体负载Pt颗粒用以糠醛选择性加氢。与高温(600 ℃)下完全煅烧获得的CeO2为载体负载Pt催化剂相比,准MOFs为载体的催化剂表现出更高的催化活性和选择性[9]。尽管准MOFs为载体负载贵金属粒子已有报道,但大都是单金属颗粒,以准MOFs为载体负载双金属颗粒有望在催化反应中,既能体现准MOFs的载体效应,又能发挥金属颗粒的合金效应。
反应底物的传质同样影响苯乙炔半加氢催化反应的性能。气态氢作为氢源被广泛应用于加氢反应,然而氢气难溶于有机溶剂,抑制了苯乙炔半加氢反应中的传质过程[10]。为此,一般采取高温、高压等苛刻的反应条件,促进氢气与液相体系中催化剂、反应底物气-固-液三相接触。最近,研究者通过串联两个独立催化反应,实现原位供氢,有效提升液相中反应底物与固体催化剂的接触,提高了传质和催化反应效率。在众多供氢剂中,氨硼烷具有高含氢量(质量分数19.4%)、输运方便和环境友好等特点,同时室温可实现氨硼烷催化脱氢,是一种理想的供氢剂[11-12]。值得注意的是,Pd基双金属催化剂不仅可催化氨硼烷脱氢[13],同时在苯乙炔半加氢反应中也具有高催化活性[14]。基于此,本文以准NH2-Ce-BDC为载体,负载PdCo纳米颗粒,制备了PdCo/准NH2-Ce-BDC催化剂,对比研究了Pd/准NH2-Ce-BDC、Co/准NH2-Ce-BDC催化剂在串联加氢反应中的性能。结果表明,PdCo/准NH2-Ce-BDC双金属催化剂在氨硼烷脱氢和苯乙炔半加氢的串联反应中表现出最佳催化性能。
1 实验材料和方法
1.1 原料
乙醇(CH3CH2OH)、氢氧化钠(NaOH)、氯化铈(CeCl3·7H2O)、氯化钯(PdCl2)、氯化钴(CoCl2),国药化学试剂有限公司;2-氨基对苯二甲酸(C8H7NO4)、氨硼烷(NH3BH3)、苯乙炔(C8H6),麦克林生物科技有限公司。
1.2 PdCo/准NH2-Ce-BDC的制备
PdCo/准NH2-Ce-BDC催化剂的制备主要分为三步,如图1所示。合成准NH2-Ce-BDC的过程如下:将550 mg NH2-BDC分散在去离子水(110 mL)中,然后缓慢滴加NaOH(1 mol·L-1)调节悬浮液pH值至5~6,随后加入484 mg CeCl3·7H2O,搅拌均匀后,静置40 min。离心分离,沉淀物用去离子水洗净,然后在70 ℃干燥12 h,得到NH2-Ce-BDC。将得到的NH2-Ce-BDC在N2气氛中300 ℃焙烧2 h,得到准NH2-Ce-BDC。
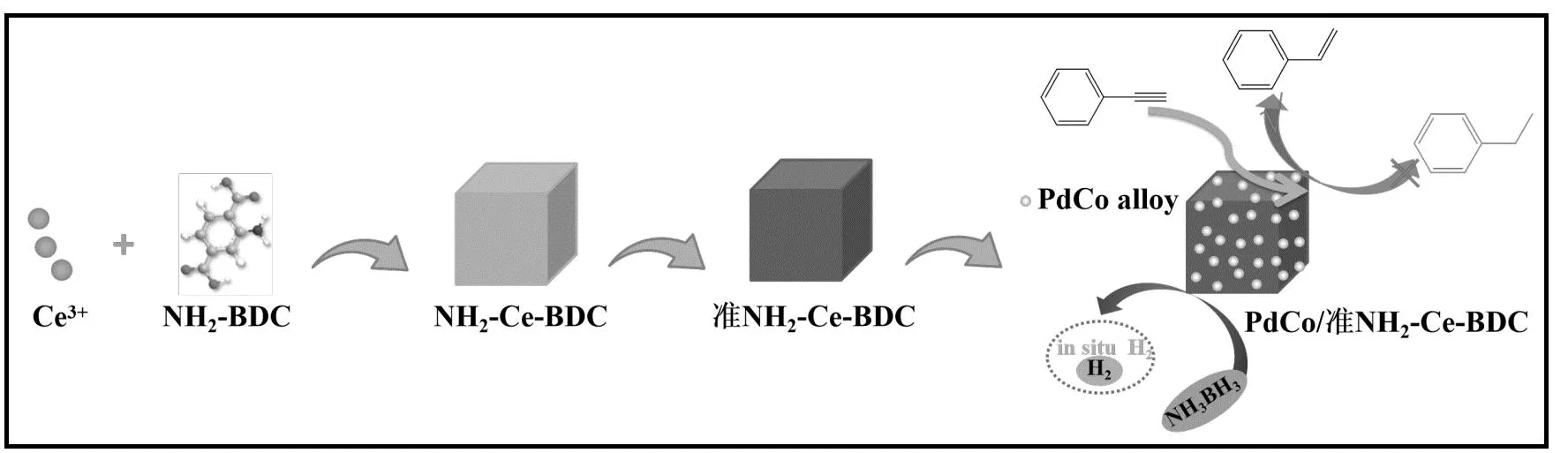
图1 PdCo/准NH2-Ce-BDC催化剂合成的示意图
通过浸渍法制备PdCo/准NH2-Ce-BDC,Pd、Co的理论负载量均为1%(质量分数)。具体负载步骤:将100 mg制备的准NH2-Ce-BDC载体分散在1 mL去离子水中,把167 μL PdCl2溶液(0.01 g·mL-1)和848 μL CoCl2溶液(20 mmol·L-1)加入上述悬浮液中,室温搅拌24 h,然后60 ℃干燥12 h。得到的固体在V(N2)∶V(H2)= 27 mL∶3 mL的气氛中250 ℃还原2 h,得到PdCo/准NH2-Ce-BDC。作为对比,用浸渍法以准NH2-Ce-BDC为载体分别浸渍Pd或Co,制备了Pd/准NH2-Ce-BDC和Co/准NH2-Ce-BDC。
1.3 分析测试仪器

1.4 催化活性实验
在双颈圆底烧瓶中,将催化剂(5 mg)和苯乙炔(50 μL)分散在6 mL乙醇中,将装满水的气体滴定管连接到反应瓶上,测量和储存反应生成的氢气。用注射器将氨硼烷溶液(0.8 mL,1 mol/L)注入混合物中,开始计时反应。反应一定时间后,用注射器提取反应混合物,离心分离催化剂。用岛津GC-MS-2020 plus(蜡柱长度:30 m,内径:0.25 mm,膜厚:0.25 mm)对反应液进行分析。首先将柱温保持在50 ℃下3 min,然后加热至250 ℃,升温速率为10 ℃·min-1,并将温度在250 ℃下继续保持3 min。用方程式(1)和(2)计算苯乙炔的转化率和苯乙烯的选择性。苯乙烯、乙苯的含量通过外标法计算获得。

(1)

(2)
2 实验结果与讨论
2.1 催化剂的结构形貌表征
首先采用FT-IR对不同样品的表面官能团进行了表征,结果如图2所示。~1 246 cm-1处的吸收峰对应于NH2-Ce-BDC中C-N的变形振动[15-16],1 544,1 421和1 382 cm-1的吸收峰归属于羧酸基团的振动[17-18]。此外,1 626 cm-1处的峰值与-NH2的N-H弯曲振动有关,而836和775 cm-1处的峰值则指认为Ce-MOFs中Ce-O键的弯曲振动[19]。显然,经过300 ℃焙烧后NH2-Ce-BDC配体未完全脱除,仍保留部分官能团。XRD表征了样品的晶体结构,并与对比样品进行了比较。如图3(a)所示,NH2-Ce-BDC的XRD谱图在5°~15°范围内出现尖锐衍射峰,与文献报道结果一致[20],这说明成功合成NH2-Ce-BDC。NH2-Ce-BDC焙烧后XRD衍射峰变宽,但没有大的位移,这可能是由于衍射峰的强度减弱所致[21]。由此可知,经300 ℃焙烧的准NH2-Ce-BDC仍保留MOFs的特征峰。负载上金属后的XRD谱图(图3(b))与准NH2-Ce-BDC相似,说明准NH2-Ce-BDC的晶型不受浸渍、还原过程的影响。此外,未观测到属于Pd、Co颗粒的特征峰,这可能与样品中金属颗粒负载量低、尺寸较小、呈高度分散态有关[22]。
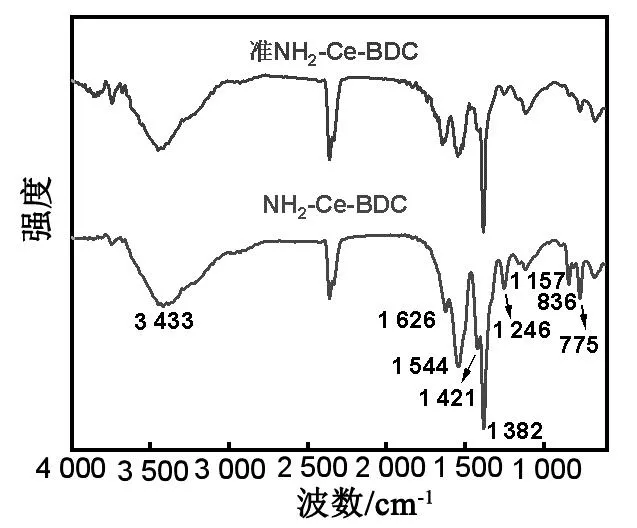
图2 NH2-Ce-BDC和准NH2-Ce-BDC的FT-IR光谱
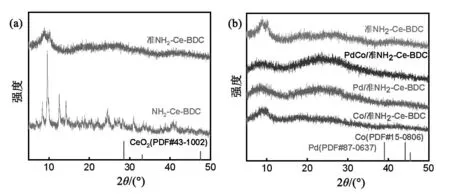
图3 NH2-Ce-BDC、准NH2-Ce-BDC、PdCo/准NH2-Ce-BDC、Pd/准NH2-Ce-BDC和Co/准NH2-Ce-BDC样品的XRD谱图
SEM和TEM表征了NH2-Ce-BDC、准NH2-Ce-BDC以及负载金属颗粒后样品的形貌。如图4(a)所示,NH2-Ce-BDC呈立方体结构,表面平整,平均棱长约为3.5 μm,焙烧后准NH2-Ce-BDC仍然保持立方体形貌,表面无明显孔出现(图4(b))。负载金属颗粒后,样品依然呈现立方体结构(图4(c)所示)。从HRTEM图像来看,0.226 和0.205 nm的晶格间距分别对应于Pd纳米颗粒(111)晶面[23]和Co纳米颗粒的(111)晶面[24],而PdCo/准NH2-Ce-BDC中0.216 nm的晶格间距处于单金属Pd(111)晶面与Co(111)晶面晶格间距之间,说明PdCo/准NH2-Ce-BDC中形成Pd-Co双金属合金结构[25]。通过元素分布图,更能清晰地看到目标样品中Pd、Co、Ce元素高度分散于立方体结构的粒子中,说明PdCo双金属粒子成功负载于准NH2-Ce-BDC。

图4 NH2-Ce-BDC(a)、准NH2-Ce-BDC(b)、PdCo/准 NH2-Ce-BDC(c)的SEM图和PdCo/准NH2-Ce-BDC(d)、 Pd/准NH2-Ce-BDC(e)、Co/准NH2-Ce-BDC(f)的 HRTEM图;(g)~(j)是PdCo/准NH2-Ce-BDC中 N,Ce,Pd,Co的mapping谱图
图5所示为样品的氮气吸附-脱附等温线和孔径分布。由图5(a)可知,所有样品均表现出IV型等温线,并在P/P0>0.6的区域内出现明显H3型回滞环,说明样品为介孔材料,同孔径分布结果一致[26]。样品的BET比表面积、孔体积数据见表1。PdCo/准NH2-Ce-BDC的比表面积为11.45 m2·g-1,略低于准NH2-Ce-BDC的比表面积,负载金属颗粒后样品的比表面积、孔隙体积和孔径分布差别不大,这说明负载金属后未破坏样品的微观结构。

图5 准NH2-Ce-BDC、PdCo/准NH2-Ce-BDC、 Pd/准NH2-Ce-BDC和Co/准NH2-Ce-BDC的 N2吸附等温线(a)及孔径分布(b)表1 准NH2-Ce-BDC、PdCo/准NH2-Ce-BDC、Pd/准 NH2-Ce-BDC和Co/准NH2-Ce-BDC的比表面积和孔体积
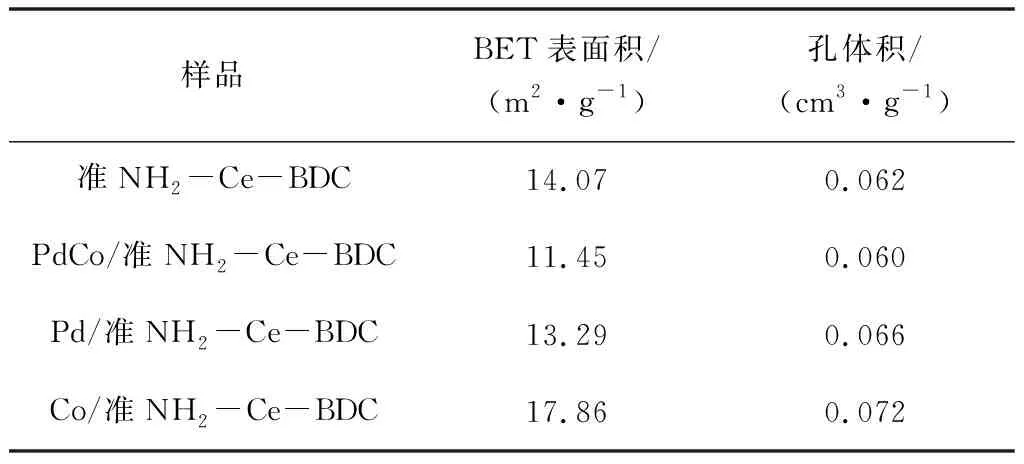
样品BET表面积/(m2·g-1)孔体积/(cm3·g-1)准NH2-Ce-BDC14.070.062PdCo/准NH2-Ce-BDC11.450.060Pd/准NH2-Ce-BDC13.290.066Co/准NH2-Ce-BDC17.860.072
形貌和结构表征结果说明PdCo/准NH2-Ce-BDC中PdCo合金化,基于此,采用XPS对PdCo/准NH2-Ce-BDC样品的元素价态进行了解析。如图6(a)所示,O 1s XPS谱图529.3和531.16 eV结合能处的峰归属于样品表面化学吸附氧(Osur)和晶格氧(Olat)[27]。Ce 3d XPS谱图包含两种Ce物种,881.6,902.8,886.1,907.54,899.6和916.6 eV处的特征峰归属于Ce4+物种,884.2和904.6 eV的峰对应于Ce3+(图6(b)),说明PdCo/准NH2-Ce-BDC载体中Ce3+和Ce4+的氧化态共存。在图6(c)中,Pd/准NH2-Ce-BDC样品Pd0的3d5/2峰在335.1 eV,掺杂Co物种后PdCo/准NH2-Ce-BDC的Pd0结合能出现正迁移,迁移量为0.5 eV,而PdCo/准NH2-Ce-BDC中Co02p3/2峰(777.9 eV)相对于Co/准NH2-Ce-BDC的Co02p3/2峰(778.4 eV)向较低的结合能偏移,这说明PdCo合金化过程中存在电子转移,Pd作为供体失电子,Co作为受体得电子,合金化电子转移过程使 PdCo/准NH2-Ce-BDC中Pd原子呈现缺电子态,缺电子态Pd活性位更有利于加氢反应[28]。
金属颗粒尺寸不同,表面吸附CO形式将会不同,因此采用CO-DRIFTS表征了PdCo/准NH2-Ce-BDC中Pd的结构,结果见图7。
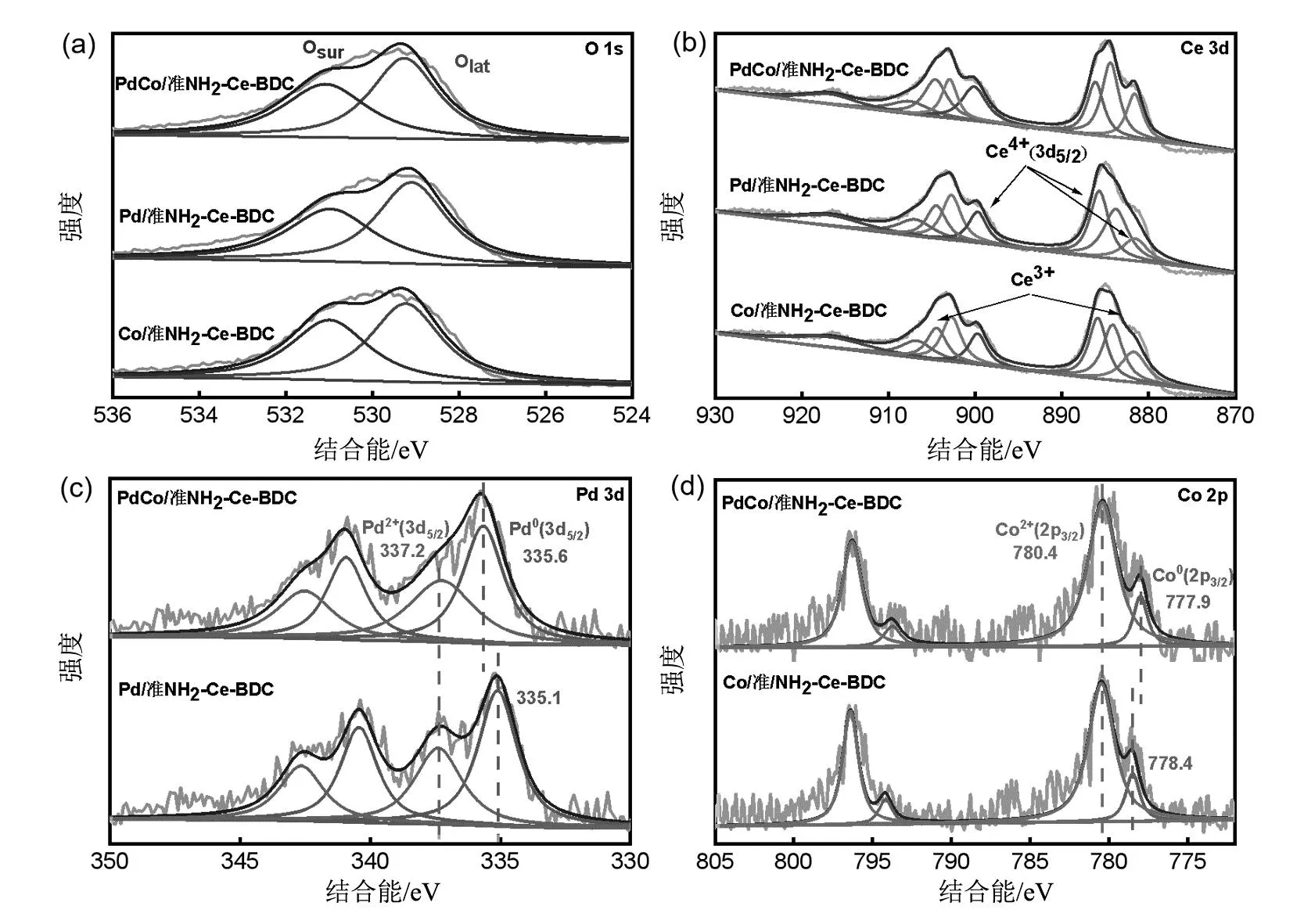
图6 PdCo/准NH2-Ce-BDC、Pd/准NH2-Ce-BDC和Co/准NH2-Ce-BDC的O 1s XPS光谱图(a)和
Ce3dXPS光谱图(b);(c)为PdCo/准NH2-Ce-BDC和Pd/准NH2-Ce-BDC的Pd3dXPS光谱图;
(d)为PdCo/准NH2-Ce-BDC和Co/准NH2-Ce-BDC的Co2pXPS光谱图

图7 PdCo/准NH2-Ce-BDC、Pd/准NH2-Ce-BDC和Co/准NH2-Ce-BDC的 CO吸附[(a),(b),(c)]及解吸光谱[(d),(e),(f)]
由图7可以看出,所有样品均出现CO的线式吸附峰(2 112,2 174 cm-1)[29],而Pd基样品的红外光谱中,在~1 890 cm-1处出现新的吸附峰,该峰归属于CO在金属Pd表面的桥式化学吸附[30],并且随着吸附时间的延长,PdCo/准NH2-Ce-BDC样品的吸附峰强度增加显著,而Pd/准NH2-Ce-BDC样品吸附峰强度变化较小,这说明CO在PdCo合金位点上的吸附态更加稳定。吸附结束后经N2吹扫脱附,CO的线式吸附峰随脱附时间增加逐渐消失,而~1 890 cm-1处的吸收峰强度随N2吹扫强度不变,这说明CO在Pd金属表面的桥式化学吸附态更稳定[31]。值得注意的是,随着脱附时间延长,在Pd负载量相同的情况下,与单金属Pd/准NH2-Ce-BDC样品相比,双金属PdCo/准NH2-Ce-BDC的CO桥式化学吸附峰强度更强,这说明双金属催化剂中PdCo合金的分散性更好[32]。
2.2 催化剂的性能
在常温常压条件下,对比研究了合成催化剂催化苯乙炔串联反应的加氢性能,结果见图8。随着反应时间的增加,苯乙炔转化率逐渐增加,PdCo/准NH2-Ce-BDC在25 min时苯乙炔转化率达到100%,苯乙烯收率为85%,然而在相同反应条件下单金属Pd/准NH2-Ce-BDC催化剂苯乙烯最佳收率仅为49%, Co/准NH2-Ce-BDC在测试条件下则无催化活性。在苯乙炔串联催化反应中,氨硼烷脱氢速率同样影响加氢活性。如图8(d)所示,对比测试了不同催化剂的氨硼烷脱氢性能,从产氢速率可以看出,PdCo双金属催化剂表现出最优氨硼烷脱氢活性,常温常压反应15 min,PdCo/准NH2-Ce-BDC催化剂产氢量是Pd单金属催化剂的1.06倍,而Co基催化剂几乎无氨硼烷脱氢活性。因此,结合上述催化剂串联反应结果可知,PdCo/准NH2-Ce-BDC催化剂中PdCo金属合金化过程中电子效应导致Pd为缺电子态,缺电子态Pd更有利于氨硼烷原位产氢和对反应底物苯乙炔的吸附[33],提高了苯乙炔串联反应加氢性能。
为了深入探究双金属PdCo/准NH2-Ce-BDC催化剂中合金效应与苯乙炔加氢反应的构效关系,将单金属Pd/准NH2-Ce-BDC和Co/准NH2-Ce-BDC催化剂物理混合后用于苯乙炔串联加氢反应,在常温常压反应条件下物理混合催化剂苯乙炔转化率为48%,苯乙烯的收率仅为47%,加氢性能远低于双金属PdCo/准NH2-Ce-BDC催化剂,这说明 PdCo/准NH2-Ce-BDC催化剂中PdCo之间的电子效应可以提升Pd的催化活性,PdCo合金化过程中 Pd作为供体失电子,Co作为受体得电子, PdCo/准NH2-Ce-BDC催化剂中缺电子态Pd更有利于反应底物的吸附活化,提高了苯乙炔的半加氢反应性能[34]。同时,PdCo/准NH2-Ce-BDC催化剂中Pd、Co双金属之间的电子效应可以抑制Co的氧化[35]。

反应条件:5 mg催化剂;50 μL反应底物;6 mL乙醇;0.8 mL AB(1 mol/L);30 ℃
在苯乙炔选择性加氢串联反应中氨硼烷脱氢作为原位氢源,与外加气态氢源相比,原位产氢更有利于活化氢物种与反应底物接触,提高传质效率。如图9(a)所示,以外加氢气(0.5 MPa)为氢源,常温常压反应25 min,PdCo/准NH2-Ce-BDC催化剂的苯乙炔转化率为100%,苯乙烯收率仅为40%,远低于串联反应中原位产氢作氢源的催化性能。在串联催化中,与外加气态氢源相比,氨硼烷原位产生的活性氢可参与苯乙炔加氢反应,提高了气-固-液三相之间的传质速率[36]以及加氢反应的催化活性。

反应条件:10 mg催化剂;100 μL反应底物;12 mL乙醇,0.5 MPa H2,30 ℃。
3 结论
以准NH2-Ce-BDC为载体,采用浸渍法负载金属,并通过H2还原等一系列步骤制得了双金属PdCo/准NH2-Ce-BDC催化剂,对比研究了其氨硼烷脱氢和苯乙炔选择性加氢串联催化反应性能,并结合XRD、XPS、CO-DRIFTS等表征手段,探明了催化剂结构与加氢性能间的构效关系。XRD和XPS等表征结果表明,PdCo/准NH2-Ce-BDC中PdCo合金活性位与载体之间具有协同效应,同时PdCo合金表现出电子效应,Pd作为供体失电子,Co作为受体得电子,合金化电子转移过程使 PdCo/准NH2-Ce-BDC中Pd原子呈现缺电子态,相比Pd/准NH2-Ce-BDC和Co/准NH2-Ce-BDC催化剂,PdCo/准NH2-Ce-BDC催化剂中缺电子态Pd合金活性位展现出最优的催化性能,在常温常压下,25 min实现苯乙炔100%转化,苯乙烯收率达到85%。对比实验指明,氨硼烷的原位产氢提高了气/固/液相接触和传质效率,更有利于串联催化反应。
猜你喜欢
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
科普创作(2018年1期)2018-11-30
石油化工建设(2018年2期)2018-07-11
电源技术(2016年9期)2016-02-27
化工进展(2015年6期)2015-11-13
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
世界热带农业信息(2014年11期)2015-01-05
自动化博览(2014年8期)2014-02-28
中成药(2014年11期)2014-02-28
