
可充放锌空电池用过渡金属氧化物双功能催化剂研究进展
2024-02-12 06:51刘渐芳赵勇智刘思佳秦运璞吴昊阳张德印贾宝瑞曲选辉秦明礼
工程科学学报 2024年1期
刘渐芳,赵勇智,王 永,刘 鸾,刘思佳,秦运璞,吴昊阳,张德印,贾宝瑞✉,曲选辉,秦明礼✉
北京科技大学新材料技术研究院,北京 100083
近年来,受现阶段严重的环境问题和日益增长的能源需求所带来的影响,开发绿色、可持续的能量转换和存储技术已经发展成为现代社会的重大课题[1-2]. 目前,锂(Li)离子二次电池作为代表性电化学能源技术,引领着储能市场,尤其在电子消费品和混合动力/纯电动汽车领域占据着主流地位. 然而,传统可充电二次锂离子电池容量一般在200~250 W·h·kg-1之间,能量密度的不足部分限制了其进一步发展和应用[3-5]. 金属–空气电池的理论能量密度比传统锂离子电池高数倍,被认为是未来社会能源储存问题的有效解决方案之一,受到越来越多的关注[6-7]. 在各类金属-空气电池中金属负极的理论比容量(质量比能量密度)、体积比能量密度和电池电压的比较中,锌-空气电池(Zincair batteries,ZABs)展现出优异的综合性能,其具有高的质量比能量密度(1218 W·h·kg-1)和体积比能量密度(6136 W·h·L-1),且锌储量丰富、价格低廉,常采用水作为溶剂,绿色环保、安全性高,因而受到人们的广泛关注[8-9].
ZABs 是一种以空气中的氧气作为正极电化学反应物质,金属锌作为负极电化学反应物质的新型储能器件,通常由四个主要部件组成[10-11]:空气电极(催化剂负载的气体扩散层)、碱性电解质、隔膜和锌电极. 放电时,锌极发生氧化反应,释放的电子通过外部电路到达空气电极. 同时,大气中的氧分子在空气电极的三相界面处发生氧还原反应(Oxygen reduction reaction,ORR),该反应产生的氢氧根离子迁移至锌电极,形成锌酸根离子,随后分解为不溶性氧化锌,总反应的平衡电压为1.66 V. ZABs 充电时,化学反应与上述过程相反,锌重新沉积在锌电极处,氢氧根离子通过析氧反应(Oxygen evolution reaction,OER)于三相界面释放氧气. 其化学反应可简化为如下所示[12]:锌阳极, Zn+4OH-⇌Zn(OH)24-+2e-,Zn(OH)24-→ZnO+H2O+2OH-;空气阴极, O2+4e-+2H2O ⇌4OH-;总反应, 2Zn+O2→2ZnO.
ZABs 在放电和充电时,空气电极分别发生ORR 和OER 反应,然而,ORR 和OER 涉及四电子和四质子的转移,动力学缓慢,往往需要较高的过电势,这制约了ZABs 的充放电性能[13],因此,高活性、高稳定性的ORR/OER 双功能催化剂是ZABs的关键材料. 目前已经被广泛研究的ORR/OER 双功能催化剂主要可以分为两大类贵金属催化剂和非贵金属催化剂,其中,贵金属催化剂主要包括用于OER 的Ir 和Ru 基催化剂和用于ORR 的Pt 催化剂. 非贵金属又可以具体分为无金属材料(如杂原子掺杂碳,富含缺陷的纳米碳材料),基于MOF 的催化剂(包括原始MOF 及其衍生物),各类非贵金属基材料(如过渡金属及其合金,过渡金属氧化物,过渡金属基复合材料,过渡金属单原子材料等)[14-18].
本文综述了不同种类的过渡金属氧化物ORR/OER 双功能催化剂,介绍了各类催化剂的活性来源及其在ZABs 能量密度、充/放电电压、稳定性等方面的表现,总结了当下研究现状中提高其催化性能的方法.
1 锰氧化物
在所有单一过渡金属氧化物基ORR/OER 双功能催化剂中,锰氧化物(MnOx)是最常见的候选材料,具有成本低、储量丰富、环境友好的优点[19].Mao 等[20]认为MnOx的ORR 催化机制可以通过以下几个步骤来描述:
ORR 遵循四电子机制,O2经过反应(1)生成HO-2,然后发生得电子还原反应(2)或者歧化反应(3)生成OH-,其中,MnOx对反应(3)具有高催化活性从而加快了整体ORR 反应速度. Roche 等[21]认为MnOx中存在大量MnIII/MnIV氧化还原电子对,可作为氧受体–供体,具体而言,首先MnO2中插入质子形成MnOOH(反应(4)),随后氧分子吸附到两个相邻的MnOOH 位点(桥接吸附位点)上(反应(5)),MnOOH 与氧气分子结合生成OH-(反应(6)),而反应(7)是Oads(吸附氧)物种的还原,该四电子反应途径可使O2还原为OH-. 同样地,OER作为ORR 的逆反应,也有研究表明MnOx催化剂的OER 活性与锰的可变价态相关[22-23].
为了研究不同晶体结构对锰氧化物催化性能的影响,Meng 等[24]制备了α 相、β 相、δ 相和无定形等不同相的MnO2材料,对比了这些材料在碱性介质中的电催化性能. 结果表明,在ORR 或者OER中MnO2的电催化活性都强烈依赖于晶体结构,并且遵循α-MnO2电催化活性>无定形MnO2电催化活性>β-MnO2电催化活性>δ-MnO2电催化活性这一趋势. 其中α-MnO2的高电催化活性可归因于以下两点:一是α-MnO2中由角与边缘共享的MnO6八面体可组成2×2 隧道结构,相比于β-MnO2的1×1 隧道结构、δ-MnO2的1×2 和1×1 隧道结构,可容纳更多的氧气分子、阳离子与水分子(如图1 所示),从而提供了更多的活性位点;二是α-MnO2具有混合价态(Mn3+/Mn4+),可通过Mn3+和Mn4+之间的氧化还原对来促进OER 过程中的电荷转移.
作为改变纳米材料电子结构的有效手段之一,金属原子掺杂也可用于提高MnOx基催化剂的活性. 已有文献报道使用Co2+、Ag+、Fe3+、Ni2+、Mo6+、Cu2+、Cr3+、V5+等不同金属离子对MnO2进行掺杂[25-27]. α-MnO2的(2×2)隧道结构是由内部存在的水分子和阳离子支撑,而该隧道结构的坍塌会引发相变,转化为β-MnO2[28]. 在低价态阳离子掺杂的MnOx中,晶格的负电荷多,可以促使溶液中更多的K+等阳离子进入到隧道中,从而提高α-MnO2结构稳定性. 而掺杂高价态阳离子会产生过量正电荷,阻碍阳离子进入隧道结构,影响α-MnO2稳定性,降低催化活性. 此外,MnO2常常表现出较差的导电性,杂原子掺杂还可加快电子传输速率,从而提高ZABs 的放/充电速率[29-31]. Bôas 等[32]制备了Bi3+和Ce4+掺杂的MnO2纳米棒,用作碱性介质中ORR 和OER 电催化时,两种催化剂的电荷转移电阻都比未掺杂样品低得多,其ORR 半波电位与10 mA·cm-2电流密度下的OER 电位(E10)的差值ΔE分别为1.07 V 和1.02 V. 其中,Ce4+掺杂MnO2纳米棒ZABs 的峰值功率密度为45 mW·cm-2,高于MnO2材料ZABs 的40 mW·cm-2,表明其良好的阴极催化作用.
除了对MnOx进行金属元素掺杂外,将其与碳、碳化物等电导率高的材料复合,也是克服其低电导的有效策略. Song 等[33]通过水热合成法在TiC纳米颗粒上沉积非晶态MnOx,该材料作为ZABs氧电极,具有高效、抗腐蚀的优点,表现出与Pt/C相当的ORR 起始电位(0.96 V)和半波电位(E1/2,0.8 V),比IrO2更低的OER 过电位(1.56 V),更低的Tafel 斜率(ORR,72 mV·dec-1;OER,110 mV·dec-1).Pandey 等[34]以次氮基三乙酸(NTA)和乙酸锰为前体,采用水热法合成了一种MnOx和氮掺杂碳复合的双功能氧催化剂(如图2 所示),其中,氮掺杂碳形成(NC)的多孔网络显著提高电催化中的电导和传质,MnOx中Mn 阳离子的混合氧化态以及碳基体中高比例的氮掺杂也有利于ORR 和OER 催化.
Chen 等[35]通过化学浴沉积方法在碳布上原位生长MnOx纳米片,所得材料表现出良好的ORR和OER 双功能活性,ORR 催化活性与Pt/C 相当,并且在10 mA·cm-2的电流密度下OER 过电位仅为388 mV. 组装而成的柔性全固态ZABs 表现优异,放电电压高达1.3 V,充电电压仅为1.9 V,可稳定运行45 h,120 次循环后的往返能量效率为62.4%.Lu 等[19]以双金属–有机骨架材料为前驱体,通过水热与煅烧合成了一种MnO/Co/多孔石墨碳复合材料,其中,原位生成的Co 纳米晶促进了碳的石墨化,提高了高电导率,异质界面也提高了OER 活性. Ji 等[36]以羧基修饰的碳纳米管作为核心前体来稳定不同金属离子,采用静电纺丝–煅烧工艺,在多孔碳纳米纤维中实现了丰富的Ni|MnO 异质界面(如图3 所示),得益于一维纤维组成的三维多孔网络结构和异种金属位点之间的强界面作用,催化剂表现出低至0.763 V 的ΔE值,由其组装而成的ZABs 具有高的开路电压、功率密度和长循环寿命. 表1 列举出了最近报导的一些锰氧化物ORR/OER 双功能催化剂的具体性能.
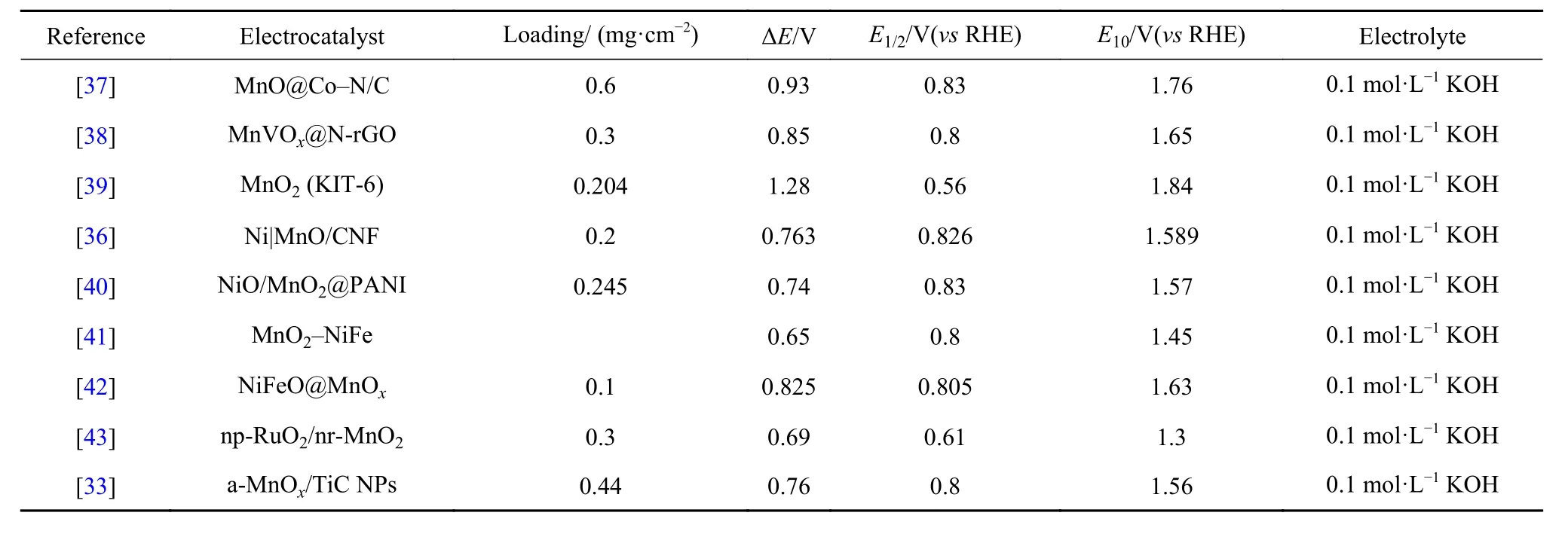
表1 一些锰氧化物ORR/OER 双功能催化剂的具体性能Table 1 Properties of some recently reported manganese oxide ORR/OER bifunctional catalysts

图3 Ni|MnO 制备流程图 [36]Fig.3 Fabrication process [36]
2 尖晶石型
尖晶石是一类化学式为AB2O4的过渡金属氧化物,其中氧离子呈立方紧密堆积,A 阳离子可以处于+2 或+4 氧化态,B 阳离子可以处于+3 或+2 氧化态,可表示为 A2+B32+O24-和A4+B22+O24-(图4(a))[44].

图4 尖晶石的(a)晶体结构示意图[44],(b) ORR 火山图和 (c) OER 火山图[48]Fig.4 (a) Crystal structure[44], (b) ORR and (c) OER volcano plots[48]
尖晶石型材料中有两种金属位点,四面体位点和八面体位点. 活性位点对氧中间体(例如OH*、O*、OOH*)吸附能被认为决定了ORR/OER 反应中的能垒和限速步骤. 对于尖晶石氧化物来说,氧中间体吸附于配位不饱和的表面金属位点上,由于八面体配位的金属离子与氧之间的相互作用更强,其被认为是氧电催化的活性位点. 具体而言,对于八面体配位的金属离子,d 轨道分裂为低能级的t2g轨道和高能级的eg轨道,当与O 成键时,eg轨道直接指向O,与O2p轨道产生大的空间重叠,导致强的化学相互作用. Wei 等[45]通过调整合成条件和金属离子的类型来调节尖晶石催化剂八面体位点金属离子的eg轨道占有率,结果表明,ORR和OER 活性与尖晶石八面体位点eg轨道占用率对应的火山图如图4(b)、(c)所示,证实尖晶石材料中的八面体金属离子是氧电催化的活性位点,其eg轨道的占有率决定尖晶石型材料的催化活性[46-47].对于四面体配位的金属离子来说,其t2g轨道和eg轨道指向明显偏离四个相邻氧的方向,产生轨道重叠较少. 尖晶石中,每个氧阴离子由一个四面体阳离子和三个八面体阳离子共享,四面体的组成会影响氧阴离子和占据八面体的阳离子之间的键合行为,当具有不同电负性的阳离子占据四面体位点时,八面体配位的金属离子以及氧阴离子上的电荷分布也会发生变化,从而对其催化活性产生影响. 对于碱性条件下的ORR/OER 反应来说,尖晶石的催化过程可以被视为沿着如图5(b)和(c)所示的反应环进行,包括氧的吸附和解吸、电子和质子的转移这四个步骤,同时伴有多价态的活性过渡金属阳离子的可逆氧化还原反应[48].其反应过程如下所示
具体来说,对于OER 而言,其反应过程可分为以下四个步骤:(1)失去电子并形成吸附原子O 和H2O;(2)OH-阴离子与吸附的O 原子反应生成吸附的OOH 物种;(3)吸附的OOH 物种进一步与额外的OH-阴离子反应形成H2O 和吸附的O2;(4)吸附的O2从催化剂表面脱离. 对于ORR 而言,其反应步骤如下:(1)OO2–/OH-置换;(2)表面过氧化物物质的形成;(3)表面氧化物的形成;(4)表面氢氧化物物质的再生[49-51].
合理改变尖晶石氧化物的组分可实现催化剂ORR/OER 活性的调控,二元尖晶石氧化物如NixCo3-xO4、MnxCo3-xO4、ZnxCo3-xO4和CuxCo3-xO4等的催化性能已经被广泛研究[52-56]. Liu 等[57]采用水热法合成ZnCo2O4/氮掺杂碳纳米管(ZnCo2O4/NCNT)复合催化剂,其在10 mA·cm-2电流密度下OER过电位为1.65 V,低于Co3O4/N-CNT 的1.67 V 和ZnCo2O4的1.69 V,具有更低的Tafel 斜率,并且其ORR 起始电位(0.95 V)和半波电位(0.87 V)都接近商业Pt/C 催化剂,优于Co3O4/N-CNT. Jiang[58]等通过引入Zn 来调整氮掺杂碳纳米管(氮掺杂碳纳米管: NCNTs, 碳纳米管: CNTs)锚定的立方尖晶石结构(ZnxNi1-xCo2O4),发现Zn0.4Ni0.6Co2O4材料的OER和ORR 活性较高. Rao 等[59]通过对Co3O4进行V掺杂,使eg占有率优化到理想的1.0,有效地平衡了氧中间体在ORR 和OER 速率决定步骤中的吸附能,作为ORR/OER 双功能电催化剂,其性能优于Pt–IrO2的组合. Wang 等[60]合成了一系列氮掺杂碳纳米管负载的钴基尖晶石氧化物电催化剂(如图6所示)(CNTs:碳纳米管),其中MnCo2O4对ORR/OER反应表现出最高的活性和耐久性,装配的ZABs 可提供827 mA·h·g-1(Zn)的比容量和74.63 mW·cm-2的功率密度,并且在300 次循环后电压没有下降.
此外,大多数尖晶石氧化物都存在导电性差和结构坚固性低的问题,有些还具有较差的O2结合和活化能力. 因此,将尖晶石氧化物与碳载体(如石墨烯、碳纳米管、碳纳米纤维和炭黑等)或其他金属进行复合,来提高材料的ORR 催化性能,并且形成导电网络促进电荷转移,也是常用的策略之一[61-64]. 尖晶石与碳载体的复合还能增强其分散均匀性,防止颗粒团聚,并提供额外的表面积.Jiang 等[65]合成出负载了Co3O4的氮和硼元素共掺杂石墨烯空心球,该材料表现出比商业Pt/C+RuO2/C更高的ORR/OER 电催化活性和耐久性. Li 等[66]合成了一种氮掺杂碳骨架负载Co3O4纳米晶的石榴状电催化剂(如图7(a),(b),(c),(d)所示),该催化剂中,小尺寸Co3O4纳米颗粒提供了丰富的活性位点,而石榴状结构碳基体能够有效防止金属氧化物团聚并提供传质通道,从而增强了催化活性和耐用性. Chen 等[67]采用静电纺丝技术将细小均匀的尖晶石钴锰氧化物纳米颗粒嵌入到氮掺杂碳纳米纤维中,形成复合催化剂. 其中,纳米纤维组成的三维导电网络同样起到了加快物质扩散、电荷转移和暴露活性位点的作用,也抑制了氧化物纳米粒子的团聚和脱离. Jiang 等[58]通过Co2+离子与Zn 有机骨架纳米片之间进行离子交换和还原反应,合成了一种多孔碳壳负载Co/Co3O4复合纳米粒子(Co/Co3O4@PGS)(如图7(e)所示),该电催化剂ORR 半波电位比Pt/C 高15 mV,在10 mA·cm-2时的OER 过电位比Ir/C 低60 mV,当集成在ZABs中时,其在10 mA·cm-2下稳定循环超过800 h. 表2总结了一些最近报导的尖晶石型ORR/OER 双功能电催化剂的性能.

表2 一些最近报道的尖晶石型电催化剂的双功能性能Table 2 ORR/OER bifunctional performances of some recently reported spinel-based electrocatalysts

图7 氮掺杂碳骨架负载Co3O4 纳米晶的石榴状电催化剂的(a,b)SEM 图像,(c)TEM 图像和(d)示意图[66];(e) Co/Co3O4@PGS 制备示意图[58]Fig.7 (a, b) SEM images, (c) TEM images, and (d) illustration of the electrocatalyst of the Co3O4 nanocrystals embedded in a nitrogen-doped partially graphitized carbon framework with a unique pomegranate-like composite architecture[66]; (e) illustration of the synthesis process for preparing interpenetrating Co and Co3O4 nanoparticles stitched in porous graphitized shells (Co/Co3O4@PGS) [58]
3 钙钛矿型
钙钛矿是一类化学式为ABO3的过渡金属氧化物,其中A 为稀土金属或碱土金属,B 为过渡金属,其中较大尺寸的稀土或碱金属阳离子占据与12 个O 进行配位的A 位点,较小尺寸的过渡金属阳离子占据与6 个O 配位的B 位点,呈立方结构(如图8(a)所示)[91]. 对于钙钛矿氧化物,一般认为占据B 位点的金属原子为其ORR 活性位点,ORR催化被认为沿着图8(b)所示的路径进行[92]. 氧空位可以增加ORR 电子转移数以减少过氧化物的形成,此外,氧空位的存在使晶格氧在钙钛矿表面移动,降低了氧迁移的屏障,使氧更容易迁移到电极材料内部[93].

图8 (a) 钙钛矿氧化物理想晶胞的示意图[91],(b)钙钛矿氧化物的ORR 催化反应途径[92]Fig.8 (a) Schematic of the ideal unit cell [91]and (b) proposed oxygen reduction reaction pathway of perovskite oxide[92]
对于钙钛矿氧化物的OER 机制,有表面吸附(Adsorbate evolution mechanism,AEM)(Me1)和晶格氧交换(Lattice oxygen oxidation mechanism,LOM)(Me2)两种机制[94]. 如图9(a)所示,AEM 的第一步是羟基(OH)吸附过程(Me1-1 步)(后缀“-1”代表反应过程的第一步,依此类推),羟基与B 位金属位点以形成相对稳定的状态(M—OH),接着,吸附物发生脱氢反应,生成表面吸附氧MO(Me1-2 步)[95].吸附氧继续与OH 反应形成M—OOH(Me1-3 步),最后,吸附物使第二个H 脱氢并形成MO=O,然后释放出氧(Me1-4 步). 由于大多数钙钛矿在Me1-2步中具有相对较高的能垒,Me1-2 步通常是其OER的速率决定步骤. 而对LOM 机制来说(图9(b)),第一步始于晶格中的氧空位(Me2-1 步),第二步中,催化剂的相邻位点吸附了一个额外的羟基M1–OH/M2–OH,称为羟基填充步骤(Me2-2 步),此步骤比AEM 中吸附氧的形成具有更低的能垒. 羟基脱氢困难形成过渡态,过渡态不稳定,最终变为M1—O=O/M2(Me2-3 步),最后释放氧,通过填充羟基恢复初始状态M1–OH/M2(Me2-4 步). 虽然LOM似乎打破了Me1-2 带来的更高能垒,但据报道只有少数高共价钙钛矿发生LOM. 除了AEM 和LOM,一些催化剂还表现出O—O 耦合机制(Me3)(图9(c)),这些催化剂对Me1-2 步骤没有高能垒,但它们的Me1-3 能垒较高. 在这种情况下,催化剂表面形成了大量的MO,且很难进一步形成M—OOH. 当MO难以转化为M—OOH 时,两个MO 会直接相互偶联(Me3-2),生成一个氧分子(Me3-3). 然而,这种机制通常很难实现,需要两个相邻的吸附氧和高能垒[96].

图9 钙钛矿八面体B 位金属位点的不同 OER 机制(红色和灰色原子分别代表氧和B 位阳离子,虚线框表示速率确定步骤[95]. (a) AEM 机制;(b) LOM 机制;(c) O—O 耦合机制.Fig.9 Different OER mechanisms of the octahedral B site in perovskite (the red and gray atoms donate oxygen and B site, respectively. The dotted box represents the rate-determining step) [95]: (a) AEM; (b) LOM; (c) O—O coupled mechanism
与尖晶石相似,钙钛矿氧化物的B 位过渡金属的电子结构也对其催化性能有着重大的影响.Suntivich 等[97]使用分子轨道理论证明,钙钛矿型催化剂的ORR 活性与B 位阳离子eg轨道的占据率呈火山型关系. 从图10(a)可以清楚地看出,在eg轨道占据率约为1 的钙钛矿,如LaMnO3、LaCoO3和LaNiO3,表现出最高的ORR 活性. eg轨道填充过少的La1-xCaxCrO3(eg占据数≈0)和eg轨道填充过多的La1-xCaxFeO3(eg占据数≈2)显示出较低的ORR 活性. 以eg轨道占据率作为描述符ORR 活性遵循Sabatier 原理,即eg轨道占用过少或占用过多会导致与含氧中间体的吸附太强或太弱,从而影响其催化活性. 在另一项研究中,他们运用相同的描述符来预测钙钛矿的OER 活性,如图10(b)所示[98],对于所测试的钙钛矿,观察到了类似的火山型相关性,Ba0.5Sr0.5Co0.8Fe0.2O3-δ钙钛矿eg轨道占据数为1.2,显示出最高的OER 活性.

图10 (a)某些钙钛矿氧化物中ORR 电流为25 µA·cm-2 的电位与eg 轨道的函数关系;(b)某些钙钛矿基氧化物中OER 电流50µA·cm-2 处的电位与eg 轨道的关系[98].Fig.10 (a) Potentials at 25 µA·cm-2 of the ORR current as a function of the eg orbital in some perovskite-based oxides; (b) potentials at 50 µA·cm-2 of the OER current as a function of the eg orbital in some perovskite-based oxides[98]
相比于其他过渡金属氧化物,钙钛矿氧化物由于其A 位和B 位均可由其他碱土、稀土金属或者过渡金属部分取代,因此钙钛矿氧化物种类更为丰富,更易调控其电子结构和催化性能. 最近报道的一些钙钛矿在碱性条件下的ORR/OER 性能已经达到甚至超过了基于贵金属(即Pt、Ir 和Ru)的催化剂的性能. 传统的固态反应合成钙钛矿氧化物往往需要长时间的高温来克服扩散障碍、形成钙钛矿晶体结构. 这种方法的主要问题是钙钛矿相的纯度不够,且由于高温团聚而造成比表面积减小. 通过结合一些新的工艺,比如溶胶–凝胶法、共沉淀水解、反胶束合成、水热法等,可以部分改善上述问题[99-103]. Cai 等[104]设计了一种固态凝胶工艺制备LaMnO3±δ钙钛矿氧化物气凝胶,其将LaMnO3±δ纳米颗粒沉积到多孔碳基质颗粒上,在高温下快速加热,碳被燃烧,形成互连的三维钙钛矿气凝胶结构体. 该材料密度低、孔隙率高、比表面积大,促进了碱性介质中的ORR 反应动力学,在0.8 V 电位下,质量活度为66.2 A·g–1,是传统块状LaMnO3的153 倍. Kuai 等[105]通过硝酸盐溶液喷雾热解法制备了一种介孔LaMnO3+δ空心微球,其工艺流程为,将含有Mn(NO3)2、La(NO3)3和嵌段共聚物的溶液通过超声波加湿器雾化,吸入管式炉后,在空气中700 ℃煅烧4 h 后得到LaMnO3+δ微球,用此方法所制备的钙钛矿微球有着尺寸小、多孔、比表面积大等优点(如图11 所示).

图11 (a)介孔LaMnO3+δ 的合成过程,(b)LaMnO3 前体SEM 图,(c~e)介孔LaMnO3+δ 产物SEM,TEM 和HRTEM 图像[105]Fig.11 (a) Synthesis procedure of the mesoporous LaMnO3+δ, (b) SEM image of the LaMnO3 precursor, and (c–e) SEM, TEM, and HRTEM images of the mesoporous LaMnO3+δ product[105]
提高钙钛矿催化活性的一种方法是调整A 位碱金属或稀土金属或B 位过渡金属阳离子种类.杂原子掺杂会造成钙钛矿中的原子和电荷重排以及价态变化,从而影响其催化活性. 对于钙钛矿氧化物的A 位掺杂,Aziz[106]等研究了不同含量的La掺杂对钙钛矿Sr2-xLaxFeO6(x=0.25、0.5 和1)的影响,发现其晶粒形态、电阻率和电化学活性都受到La掺杂的强烈影响,其中Sr1.5La0.5FeO6在0.1 mol·L-1KOH 碱性溶液中表现出最优异的ORR 和OER 电化学活性. 不同A 位碱金属的掺杂会改变钙钛矿的八面体堆叠构型,从而对钙钛矿催化活性造成影响. Zhao 等[107]合成了一系列具有相似纳米颗粒形态和比表面积的钙钛矿AMnO3(A = Ca、Sr 和Ba),研究结果表明,A 位元素的离子半径变化会导致不同的八面体堆叠,随着离子半径的增加(Ca、到Sr 到Ba),钙钛矿AMnO3发生从正交结构到六方结构的相变(图12). CaMnO3呈正交结构,由共角八面体组成;SrMnO3含面共享和角共享八面体,交替以立方和六方形式堆叠;BaMnO3具有六方结构,仅含有共面八面体. 具有立方和六方混合堆叠构型的SrMnO3表现出最好的ORR 催化活性:半波电位SrMnO3(0.81 V)>半波电位CaMnO3(0.78 V)>半波电位BaMnO3(0.74 V),作为ZABs 催化剂,SrMnO3显示出与Pt/C 相当的峰值功率密度(~233 mW·cm-2),以及400 h 的充放电循环稳定性.

图12 不同的八面体堆叠结构[107]Fig.12 Different octahedron stacking configurations[107]
钙钛矿的B 位原子常常采用Fe、Co、Ni 和Mn等其他过渡金属进行部分取代,用于调控它们的OER电催化性能. Hua 等[108]的研究表明,在PrBaMn2O5+δ双钙钛矿中引入适量的Fe,能使B 位阳离子达到高价态和最佳的eg轨道填充,有助于获得更好的ORR/OER 电催化性能. Wang 等[109]报道了一系列Mn掺杂的La0.8Sr0.2Co1-xMnxO3钙钛矿,其中La0.8Sr0.2Co0.4Mn0.6O3用作ZABs 的阴极电催化剂时,比Pt/C具有更好的性能和循环耐久性,其优异活性归因于掺杂导致的B 位阳离子化学价的变化,其Co3+/Co2+比值满足eg占据接近1 的原则. 非金属元素的掺杂,特别是磷,已被证明可有效提高碱性条件下钙钛矿氧化物的ORR 性能,例如,Shen 等[110]以金属硝酸盐、醋酸盐和NH4H2PO4为原料,采用溶胶–凝胶法制备了磷掺杂的La0.8Sr0.2MnO3-δ,其结构中5%(原子数分数)的磷的存在增加了其比表面积并增强了其对含氧中间体的吸附性,如图13(编号中P 后数字代表P 的掺杂量)所示未掺杂的Mn 2p 3/2 和Mn 2p 1/2 的特征峰之间的分离距离约为11.7 eV;随着P 掺杂含量的增加,两个峰的位置逐渐向更高的结合能移动表明在更高的掺杂含量下Mn 的氧化态增加,材料内新增了Mn3+. 相比于未掺杂的材料,Mn 的这种混合价态促进电荷转移到吸附氧的过程,从而通过快速氧化还原对实现高ORR 活性,起始电位为0.93 V.

图13 (a) LSM、LSMP0.02、LSMP0.05 和 LSMP0.1 的 Mn 2 p XPS 光谱; (b) LSM、LSMP0.02、LSMP0.05 和 LSMP0.1 的解析 Mn 2 p XPS 光谱[110]Fig.13 (a) Mn 2p XPS spectra for LSM, LSMP0.02, LSMP0.05, and LSMP0.1 and (b) resolved Mn 2p XPS spectra for LSM, LSMP0.02, LSMP0.05,and LSMP0.1[110]
提高钙钛矿催化性能的另一种方法是引入氧空位. 理想的ABO3钙钛矿结构在其A 位和B 位包含等量的阳离子(即A/B 阳离子比=1). 但是,在许多情况下,当A/B 阳离子比显著偏离1 时,钙钛矿晶格结构仍然可以稳定,这是由于额外氧空位的存在. Mefford 等[111]指出,La1-xSrxCoO3-δ钙钛矿中更多的氧空位使Co—O 键更强共价性,提高了OER 活性. Gui 等[112]设计了一种Ce0.9Gd0.1O2-δ修饰(Pr0.5Ba0.5)CoO3-δ钙钛矿复合催化剂,与原始(Pr0.5Ba0.5)CoO3-δ催化剂相比,更多的表面氧空位使其具有更优异的双功能活性以及耐久性. Yan 等[113]发现引入A 位阳离子缺陷能够显著提高LaFeO3(LF,L后数字代表La 的含量)的ORR 和OER 电催化活性,其活性的提高可归因于表面氧空位和少量Fe4+物质的产生. 从图14(J:电流密度)可以看出,在1.63 V时,A 位阳离子缺乏的La1–xFeO3-δ催化剂比原始LF催化剂具有更好的质量活性和比活性,L0.95F 的Tafel 斜率最小.

图14 (a) LF、L0.98F、L0.95F 和L0.9F 催化剂在1.63 V 时的OER 质量活性(MA)和比活性(SA); (b)LF、L0.98F、L0.95F 和L0.9F 催化剂的Tafel 图 [113]Fig.14 (a) OER mass activity (MA) and specific activity (SA) of the LF, L0.98F, L0.95F, and L0.9F catalysts at 1.63 V; (b) Tafel plots of the LF, L0.98F,L0.95F, and L0.9F catalysts[113]
高导电性对于提高钙钛矿的ORR/OER 电催化性能至关重要. 大多数钙钛矿导电性较差,将其与高导电碳材料(如碳管、石墨烯)复合是一有效策略. 例如,Guo 等[114]报道了一种由(PrBa0.5Sr0.5)0.95Co1.5Fe0.5O5+δ和多孔氮掺杂石墨烯复合的ORR/OER催化剂;Hu 等[115]通过水热法制备了LaNiO3纳米棒/石墨烯复合催化剂,作为ZABs 的双功能催化剂,充放电的循环性能和稳定性显著高于商用Pt/C.Prabu 等[116]将一维钙钛矿LaTi0.65Fe0.35O3-δ(LTFO)纳米颗粒嵌入在碳纳米棒表面和内部,凭借高表面积、高氮官能团的、高度石墨化碳的存在以及高活性 LTFO 中心在高导电碳纳米棒上的良好分散,其表现出了良好的双功能活性(ΔE=1.1 V). 表3总结了最近报导的部分钙钛矿双功能ORR/OER催化剂的性能.

表3 一些钙钛矿型电催化剂的双功能性能Table 3 Bifunctional properties of some perovskite-type electrocatalysts
4 总结及展望
作为一种新型的储能技术,可充放电锌–空气电池(Zinc-air batteries,ZABs)因其高的能量密度、高安全性和低成本,被认为具有广阔的应用前景.但是,若要实现锌空气电池的商业化应用,尚有大量的科学问题有待解决. 其中,ORR 及OER 的缓慢反应动力学制约了ZABs 的充放电性能,因此,目前迫切需要开发一种高活性,高稳定性的催化剂. 本文对最近报导的具有ORR 和OER 双功能活性的过渡金属氧化物电催化剂的研究进展进行了总结并对其进行如下展望.
(1)要对ORR 和OER 的反应过程有更深入的了解,包括反应途径、活性位点的识别、界面行为等方面,同时需要开发更先进的表征方法,相关催化机制的日益清晰将对新型催化剂的开发带来更多帮助.
(2)双功能催化剂在ZABs 工作时的位点失活问题需要格外关注,特别是ORR 活性位点在OER条件下易发生不可逆的损坏,这往往给材料的复合设计带来困难. 目前已有新的电池设计的报道,将ORR 和OER 解耦,分别在放电电极和充电电极上各自发生,目的就是避免上述问题.
(3)碳复合仍然是目前双功能催化剂主要的制备策略之一,一是因为碳材料可以增加电化学面积,并提高导电性,同时碳材料也容易形成多孔结构来加快传质;此外,碳基材料,尤其是碳基单原子材料本身有较高的ORR 活性,将其与OER 活性物质复合后容易展现出双功能特性. 但是,随之而来的,碳在OER 条件下的氧化和ORR 位点失效问题需要进一步解决.
(4)总的来说,由于ORR 和OER 的四电子特点,目前报道的各类催化剂仍然具有很高的过电势,导致可充放电ZABs 能量效率仍然较低,这极大限制了该类电池的广泛应用,因此,发现新的高活性催化剂十分必要且任务艰巨,人工智能、机器学习等大数据技术未来有望带来新的机遇.
猜你喜欢
山东冶金(2022年4期)2022-09-14
耐火材料(2022年4期)2022-08-28
中国宝玉石(2022年2期)2022-04-25
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
中国有色金属学报(2018年2期)2018-03-26
物理学进展(2017年1期)2017-02-23
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
中国洗涤用品工业(2015年7期)2015-02-28
太阳能(2015年4期)2015-02-28
太阳能(2015年2期)2015-02-28
