
高含卤阻燃剂污染特征及分析方法研究进展
2024-02-29 02:17张文睿辛正豪曾冬娟孙鹏飞孔彪梁鹏
科学技术与工程 2024年3期
张文睿, 辛正豪, 曾冬娟, 孙鹏飞, 孔彪, 梁鹏
(1.山东科技大学安全与环境工程学院, 青岛 266590; 2.山东科技大学化学与生物工程学院, 青岛 266590)
卤代阻燃剂(halogenated flame retardants,HFRs)被广泛添加于各种塑料制品、建筑材料、电子电器设备、家居产品与纺织品等中,通过增加材料耐燃性,阻止或延缓材料的燃烧[1]。由于HFRs多作为添加型阻燃剂使用,其与产品基体缺少化学键束缚,易在产品生产、使用及回收处置等生命全周期过程中释放到环境中[2-3],并长时间滞留难以降解[4]。HFRs可经大气呼吸、灰尘摄入和皮肤接触等途径进入人体;除此之外,进入环境中的HFRs具有持久性及生物富集性,可经食物链传递进入人体,干扰人体内分泌系统[5],并带来潜在的神经发育毒性[6]。HFRs造成的污染及其健康效应是近20年来环境学界持续关注的前沿热点,其中产用量最大的多溴联苯醚(polybrominated diphenyl ethers, PBDEs)等已被列入受国际监管和限制的《关于持久性有机污染物的斯德哥尔摩公约》(POPs公约)名单,被逐步淘汰禁用[7-8],这也促使种类繁多的“理想”阻燃替代品的诞生。其中,高含卤阻燃剂(highly halogenated flame retardants, HHFRs)因含卤量高,阻燃性能优越,性价比突出,作为阻燃剂市场的宠儿而得到广泛应用,最典型的HHFRs包括十溴二苯乙烷(DBDPE)、得克隆(DP)、四溴双酚A双(2,3-二溴丙基)醚(TBBPA-DBPE)、四溴双酚S双(2,3-二溴丙基)醚(TBBPS-DBPE)、三(2,4,6-三溴苯氧基)-1,3,5-三嗪(TTBP-TAZ)、三(三溴新戊基)磷酸酯(TTBNPP)等。
相比低卤取代HFRs,HHFRs具有更大的分子体积,早期曾被认为不易在生物体内蓄积[9],因此,相关的管控法规严重滞后。越来越多研究证据显示,HHFRs易在光、热和生物酶作用下降解为低卤转化产物等[10],从而带来更显著的毒性效应[11]。然而,目前对于HHFRs环境行为及归趋的认识相对非常有限,这与HHFRs大量的生产使用现状不符。另一方面,由于HHFRs具有沸点高、挥发性差及高温易脱卤降解等特性,故以往对常规低卤代HFRs的分析方法无法适用于HHFRs的分析测定。例如,使用气相色谱(gas chromatography,GC)分析HHFRs需要较高的进样口温度及色谱柱温度,而针对其高温易裂解的特点则需要缩短其在高温条件下的停留时间,即要求仪器分析时间尽可能短,这与难挥发特征不相符,给准确定性定量带来一定的挑战。因此,当前针对HHFRs定性定量的研究主要偏重于色谱质谱分离检测条件、色谱柱和离子源等参数的优化,从而提升分析方法的灵敏度及检测结果的可靠性。在此,现针对十溴联苯醚(BDE-209)、十溴二苯乙烷(DBDPE)、得克隆(DP)、四溴双酚A/S衍生物(TBBPA/S-DBPE)、三(2,4,6-三溴苯氧基)-1,3,5-三嗪(TTBP-TAZ)及三(三溴新戊基)磷酸酯(TTBNPP)等几种典型HHFRs的理化性质、环境赋存、前处理方法及仪器检测方法进行综述,以期为开展HHFRs及其转化产物的分析提供参考。
1 HHFRs的理化性质
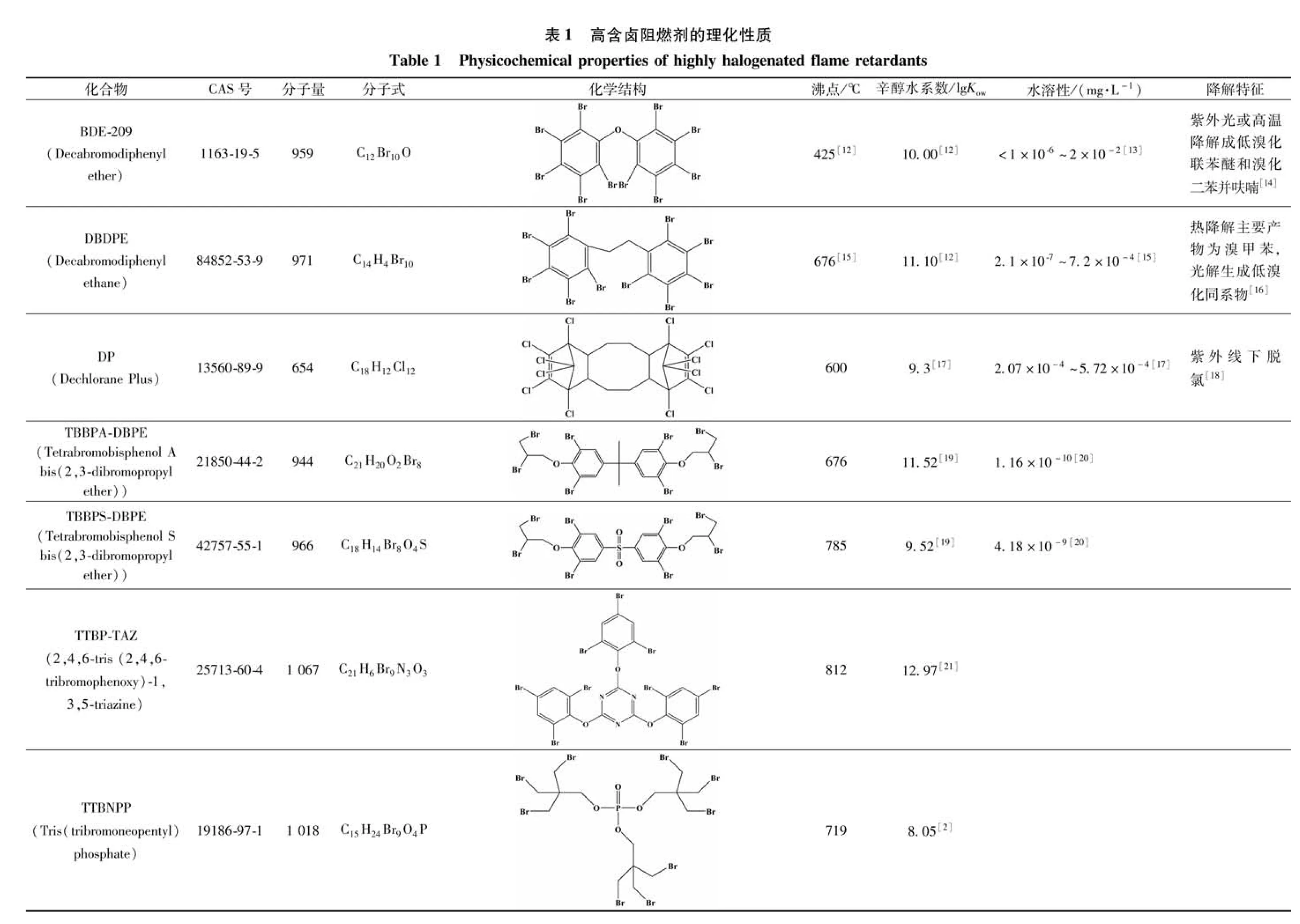
表1中列举了包括BDE-209、DBDPE、DP、TBBPA/S-DBPE、TBBP-TAZ、TTBNPP等7种典型HHFRs的基本理化性质。这些HHFRs相对分子质量介于654~1 067,其中含氯取代的DP分子量小于700,其他的分子量均在700以上。含溴取代的HHFRs有8~10个不等的溴原子,含溴量在66.2%~83.3%;而DP则有12个氯原子,含氯量为65.1%。在上述7种HHFRs中,BDE-209的沸点最低(425oC),而其他的HHFRs沸点均高于BDE-209,BDE-209高沸点难气化的特性已有共识,可以预见这些沸点更高的HHFRs将更难气化。此外,HHFRs辛醇水分配系数(Kow)介于8.05~12.97,表明这类化合物具有较强的疏水性,即微溶或不溶于水,从而表现出在水环境中易在沉积物蓄积的特征。由于Kow>4.5的化合物具有更高的生物累积潜力[2],这些HHFRs在水生生物体内蓄积可能带来潜在的生态及健康风险。除此,大部分HHFRs易在紫外光或高温下降解,如BDE-209可在紫外光条件下降解成低溴取代联苯醚和溴代二苯并呋喃[14];DP在紫外线照射下会发生脱氯等[18],这给HHFRs特别是脱卤产物的分析带来一定的干扰,需更可靠的分析方法来清晰判断脱卤产物来源。
2 HHFRs的环境赋存
HHFRs的高疏水性使其极易吸附于有机碳或附着于大气颗粒相中,并通过干湿沉降汇集入灰尘、土壤及沉积物中[13,17]。此外,大气-植物交换是HHFRs从大气环境向陆地生态系统转移的重要途径[16],且由于HHFRs具有亲脂性,会积聚在富含脂质的物质中,并在食物链中被生物放大[17],因而易在动植物体内富集。普通人群主要通过室内灰尘、食物、大气颗粒物摄入暴露于HHFRs[18]。因此,现有的研究大多关注HHFRs在土壤、沉积物、灰尘及大气颗粒物等环境介质,以及动植物与人体内的赋存特征。表2中列举了部分国内外相关研究中HHFR在环境介质及生物体内的赋存浓度水平。
2.1 环境样品
通过部署在全球范围的大气被动采样(global atmospheric passive sampling,GAPS)网络,Rauert等[25]发现BDE-209在16%的大气样品中检出,浓度范围在0.8~52 pg/m3,其中BDE-209在北美地区的浓度最高,而它在欧洲地区的浓度均低于检出限。相较于全球范围的研究,Zhao等[26]调查了中国十个省会城市大气中多种HFRs,发现BDE-209与DBDPE是溴化阻燃剂中占比最高的物质(>80%)。其中,BDE-209在颗粒相中的浓度介于未检出到1.82×103pg/m3,在气相中的浓度(未检出到34 pg/m3)相对较低,且冬季大气中BDE-209水平[(63±146) pg/m3]远高于夏季[(15±27) pg/m3]。该研究报道的大气BDE-209浓度水平与全球研究(28 pg/m3)相似[25];而大气中DBDPE的浓度范围在1~1 141 pg/m3,也呈显著的季节变化特征。
作为人体暴露HHFRs的主要来源之一,灰尘样品中也检出了高含量的HHFRs,这些灰尘主要采集自家庭或电子垃圾拆解车间等室内场所。BDE-209作为最早一批投入使用的HHFRs,大规模的使用已导致其在环境或生物介质中普遍存在。Harrad等[24]在2008年报道了来自新西兰、英国、加拿大、美国共计78个家庭室内灰尘样品的PBDEs浓度,其中BDE-209的中位浓度为:英国(2.8×103ng/g)>美国(1.3×103ng/g)>加拿大(5.6×102ng/g),新西兰作为从未直接生产或进口PBDEs的国家,在其室内灰尘样品中虽未检出BDE-209,但仍检测到三至六溴联苯醚的存在,说明含PBDEs商品的国际贸易是影响其全球分布的重要途径。此外,在英国的两个样品中BDE-209检测出了1×105ng/g和5.2×105ng/g的极高浓度,表明一些英国个人暴露于BDE 209的水平远远超过了通常通过饮食获得的水平。除BDE-209外,Ballesteros-Gomez等[29]在荷兰居民室内灰尘检出TTBP-TAZ的浓度范围为20~2.22×104ng/g。Guo等[31]在北美某电子垃圾回收地灰尘及大气样品中均检出TTBP-TAZ,浓度范围分别为1.17×103~4.2×104ng/g和2.23~10.3 ng/m3;同时灰尘中检测到BDE-209、DBDPE和TBBPA-DBPE的中位浓度分别为7.9×104、1.44×103、1.58×103ng/g,TTBP-TAZ在室外环境样品中均未检出,说明其主要来自电子垃圾回收活动。此外,样品中TTBP-TAZ与BDE-209等其他阻燃剂无显著相关性,表明它们具有不同的来源或用途。相较于国外数据,Peng等[27]报道了上海居民住宅灰尘样品中BDE-209浓度(范围:70~1.01×104ng/g)居于全球研究中位水平,而DBDPE浓度(范围:100~9.5×103ng/g)高于多数国家或地区。Shen等[30]在中国华南地区某电子垃圾拆解车间及当地住宅灰尘样品中均检出TTBP-TAZ,浓度范围分别为nd~374 ng/g干重、4.6~431 ng/g干重。
沉积物是水生态系统中HHFRs的重要储藏库,对于深层沉积物的研究亦可揭示HHFRs的时间趋势。Yang等[32]调查了美国五大湖地区沉积物中多种HFRs,其中BDE-209检出率为83%,浓度范围为0.87~106 ng/g干重;DBDPE检出率为46%,浓度范围为0.11~2.8 ng/g干重。Cai等[36]在北极西部的沉积物检测到BDE-209、DBDPE的浓度范围分别为nd~805 pg/g干重、nd~453 pg/g干重。Hoang等[35]检测采集自日本西南部Beppu Bay的沉积物柱状样,其中BDE-209在柱状样0~15 cm处被检出,浓度范围为4.9×103~3.14×104pg/g干重,在8~9 cm处浓度达到峰值;在1991—2011年间采集的沉积层柱状样检出DBDPE,浓度范围为69~850 pg/g干重,在此期间 DBDPE 与 BDE-209 的浓度比值逐渐增加,表明这两种化合物产用量呈相反趋势,亦证明DBDPE作为BDE-209替代品而存在。近年来,众多发达国家将重工业和制造业工厂迁移至发展中国家[44],由此也将污染源转移到了亚洲、非洲等地区。Zhu等[33]发现DBDPE是珠三角表层沉积物主要污染物(范围:1.52~1.714×103ng/g干重),并检出DP浓度范围为未检出~9.46 ng/g干重,此外,该团队还调查了中国长三角地区海底及河流沉积物样品中常规及新兴卤代阻燃剂浓度,测得BDE-209、DBDPE、DPs的浓度范围分别为0.038~10.7、0.176~19、0.591~7 ng/g干重[34],为珠三角及长三角地区水生态系统的HHFRs污染特征提供了依据。中国江桂斌院士及其团队始终致力于推进环境新污染物领域的研究,该团队Xie等[45]首次在亚洲菲律宾东北部马里亚纳海沟等深渊海沟沉积物样品中检测到BDE-209和DBDPE,检出频率为100%,且该研究发现BDE-209为16种PBDEs(范围:179~1 220 pg/g干重)中的主要同系物,DBDPE为9种新型目标BFRs(brominated flame retardants;范围:244~1 250 pg/g干重)中检出的主要污染物;此外,Liu等[37]首次在中国莱州湾溴化阻燃剂工厂附近土壤检出TBBPA-DBPE,浓度范围在nd~1.3×107ng/g范围,并证实醚键断裂和脱溴是TBBPA衍生物形成的主要途径。此外,工业污水的排放也是HHFRs重要的污染来源,TTBNPP化合物在日本十溴联苯醚处理设施及其下游污水处理厂的污水样品中被检出[3],浓度范围为nd~450 ng/mL,有必要进一步结合生态毒理学数据来揭示其环境风险。
2.2 生物体
由于HHFRs具有亲脂性,而大气-植物交换可使HHFRs从大气环境向陆地生态系统转移。鉴于松针具有较高的比表面积和脂质含量,易于捕获HHFRs,因此Jia等[22]以松针为“被动监测器”监测上海大气中的HHFRs。测得松针样品中BDE-209、DBDPE和DP的浓度范围分别为nd~324、nd~2.81、8.15×10-4~1.09 ng/g。Wang等[38]在中国南方某电子垃圾拆解地附近河岸植物样品检出BDE-209,浓度为3.32~76.3 ng/g干重,高于周边农田植物样品BDE-209浓度(14.97~18.15 ng/g),该发现表明,电子垃圾拆解活动释放的BDE-209通过直接排放、排气和干/湿沉积转移到水生和陆地生态系统中,导致其在周边植物体内富集。作为PBDEs替代品,DBDPE也在哈德逊湾和格陵兰的北极熊及其亚种群生物组织样本中检出[46-47],HHFRs等新有机卤素污染问题仍将是北极生态系统需要持续关注的环境问题。银鸥作为五大湖水生生态系统中污染物负荷的重点关注物种已有多年历史,Smythe等[39]调查了美国五大湖地区银鸥生物组织样品,测得BDE-209在脂肪样品中浓度水平最高[范围(623±144) ng/g脂重],其次为肌肉样品[范围(38.3±11.9)ng/g脂重]、脑组织样品[范围(16.4±4.9) ng/g脂重]和肝脏样品[范围(8.9±3.4)ng/g脂重]。Gauthier等[40]检测了美国五大湖地区银鸥蛋、反刍物及粪便样品中的TBBPA-DBPE浓度,从上述各类样品中得到TBBPA-DBPE浓度范围分别为nd~497 ng/g湿重、nd~21.7 ng/g干重、nd~16.3 ng/g脂重,TBBPA-DBPE暴露与陆地食物来源相关。此外,在其早期研究中检测到五大湖地区银鸥蛋存在TBBPS-DBPE,但浓度低于定量限(0.3 ng/g湿重)[48]。此外,Liu等[19]检测了渤海生物样品中的TBBPS-DBPE的浓度,浓度范围为nd~55.5 ng/g脂重,HHFRs在海洋食物链中的富集风险同样值得关注,海产品摄入同样是人类暴露于某些HHFRs的主要途径。
2.3 人体赋存
尽管分子体积较大,在国内外诸多人体样品中也发现了HHFRs的赋存。Gao等[41]在对健康人群血清中的阻燃剂浓度水平检测中测得BDE-209、DBDPE的浓度为nd~30、nd~43.9 ng/g,研究表明健康人群在日常生活中亦暴露在高水平的HHFRs下。Liang等[42]比较了浙江温岭电子垃圾拆解工人与非职业居民以及城市居民头发中的阻燃剂浓度水平,结果表明电子垃圾拆解工头发和血清样本中的BDE-209(46.2~724、124~2.14×103ng/g)、DBDPE(21.2~239 ng/g、26.7~440 ng/g)浓度显著高于非职业居民(BDE-209:nd~176 ng/g、8.9~199 ng/g;DBDPE:nd~197 ng/g、4.2~127 ng/g)和城市居民(BDE-209:nd~35、nd~14.6 ng/g;DBDPE:nd~36.6 ng/g、nd~33.2 ng/g)。BDE-209是电子垃圾地区人体头发和血清样本含卤阻燃剂中的主要污染物,而DBDPE 是市区样本含卤阻燃剂中的主要污染物。Wang等[43]调查了中国山东某BDE-209生产厂和某DBDPE生产厂工人血清和尿液样品中HFRs。结果发现BDE-209厂工人血清中BDE-209浓度为202~5.71×104ng/g脂重,尿样中BDE-209中位浓度为1.12 ng/mL;DBDPE厂工人血清中DBDPE浓度为87~5.44×104ng/g脂重,尿样中DBDPE中位浓度为8.6 ng/mL,说明生产阻燃剂职业人群更易暴露于HHFRs的污染,这一结论与Matsukami等[49]和Ma等[50]的研究结果相吻合。
目前针对TTBNPP在环境及生物样品中的检出还缺乏相关报道,尚有待分析领域科研工作者加强相关研究。
3 HHFRs的样品前处理
如前所述,HHFRs在污染源区域,如阻燃剂生产工厂及电子垃圾拆解回收地等地区的环境样品中检出水平较高,而在背景环境样品中的浓度相对较低。因此,要对环境及生物样品中单种或多种HHFRs进行准确定性定量分析,必要的前处理不可或缺。由于大部分HHFRs在紫外光作用下易脱卤,因此在前处理过程中宜采用棕色的玻璃器皿进行避光处理;由于HHFRs容易吸附在各种容器及颗粒物表面造成回收率损失,故需要加入相应的内标和回收率指示物作质控。对于沉积物和土壤样品,在样品提取之前需进行干燥、均质和过筛,并除掉硫化物的干扰;对生物样品中痕量HHFRs的分析,在去除样品中所含脂质的同时,需特别注意实验室可能存在的高污染本底值的干扰。
3.1 环境样品
环境基质中的HHFRs的提取、浓缩及净化与其他有机污染物类似,如图1(a)所示,样品提取方法包括索氏抽提、超声萃取、加速溶剂萃取(ASE)等,ASE和超声萃取相较于索氏提取具有萃取时间短、溶剂消耗少、提取效率高等优点。在对不同环境样品的提取净化中,常采用超声萃取+固相萃取(solid-phase extraction,SPE)联合,使用不同比例的正己烷/丙酮/二氯甲烷等混合溶剂萃取目标物,而后使用Florisil®柱、浓硫酸硅胶柱等作净化处理[51-53]。如Ali等[51]使用超声萃取+固相萃取的方法对50 mg灰尘中包含BDE-209、DBDPE在内的多种HFRs等进行提取净化,先采用硅胶柱将超声提取液中不同极性组分的HFRs进行分离,对含有BDE-209和DBDPE的组分进一步采用浓硫酸硅胶柱(44%)和弗罗里硅土(Florisil)柱净化,该方法得到DBDPE、TBBPA-DBPE的回收率分别为94%和48%,方法检出限均为6 ng/g。对于气体样品和大气颗粒物样品的浓缩净化,通常采用索氏提取加SPE的方法。譬如,Zhao等[26]采用二氯甲烷对聚氨酯泡沫(polyurethane foam,PUF)和石英纤维过滤器(quartz fiber filter,QFF)所采集的气体样品和大气颗粒物样品进行24 h索氏萃取,提取液浓缩后再经多层硅胶柱净化得到含BDE-209、DBDPE的萃取物。此外,Liu等[54]开发了基于薄层色谱(thin-layer chromatography,TLC)对高污染土壤样品中多种TBBPA/S及其转化产物的富集方法,该方法可经过硅胶GF254预涂TLC对土壤提取液进行分离富集,再将分离后的样品点进行溶解后分析。与传统的预处理技术相比,TLC分析时间更短、提取和净化步骤更快捷。此外,Suzuki等[3]将从水和污泥样品中得到的粗提物进行二甲基亚砜-正己烷分馏萃取,再将二甲基亚砜馏分与氯化钠溶液混合,用乙酸乙酯萃取,得到含有TTBNPP的萃取液。
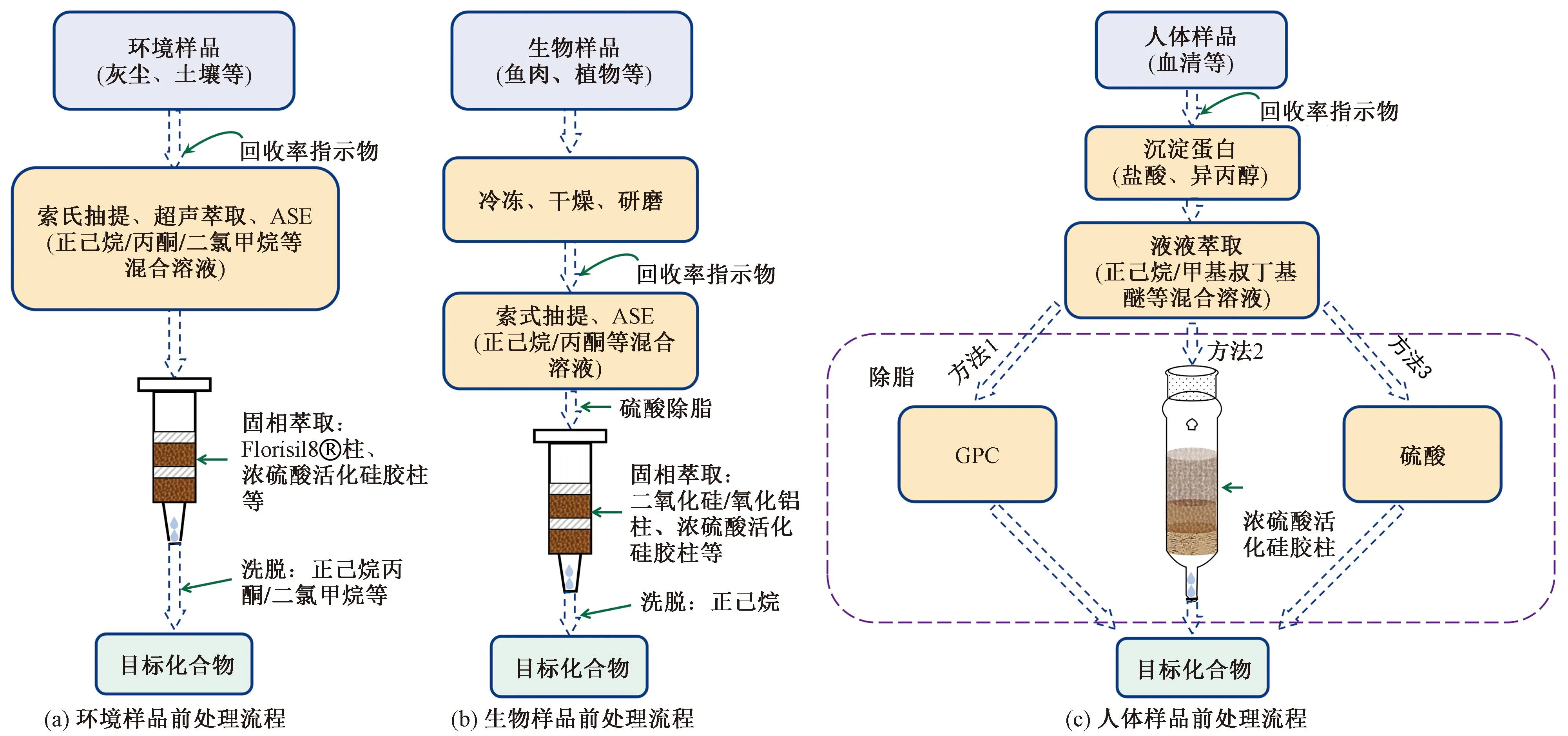
图1 样品前处理流程Fig.1 Sample pretreatment process
3.2 生物样品
如图1(b)所示,常见的生物样品主要有生物组织和植物等。这些固体样品通常需先冷冻干燥除水,经粉碎均质,再经索氏抽提或ASE提取。Wang等[38]将5 g植物样品与Na2SO4混合,在索式装置中用200 mL正己烷/丙酮(体积比1∶1)混合溶液萃取8 h,将提取物旋转蒸发至干,并用10 mL正己烷重新溶解,旋蒸溶解过程重复3次,然后用一定量的浓硫酸除脂,收集有机相,通过多层硅胶柱浓缩至1 mL,测得阻燃剂总回收率在78.5%~117.3%,其中BDE-209检出限为0.5 ng/g干重。Babut等[55]采用ASE通过甲苯/丙酮(体积比70∶30)混合溶液提取鱼肉样品中目标物,溶于正己烷,采用SPE的方法将正己烷提取物通过多层酸性硅胶柱,用正己烷洗脱后,得到的阻燃剂总回收率在65%~115%。此外,Cloutier等[56]针对鱼肉和贻贝组织样品开发了一种基于快速、简单、便宜、有效、坚固耐用、安全(QuEChERS)萃取和凝胶渗透色谱(gel permeation chromatography,GPC)、SPE净化的方法——将6 g冷冻干燥均质后的样品与5 mL水在50 mL聚丙烯锥形管中涡旋混匀,加入10 mL乙酸乙酯后手摇1 min,然后加入盐包(2 g NaCl和4 g Na2SO4)再手摇1 min,经离心后收集上层提取液;向管中继续加入10 mL乙酸乙酯手摇1 min,经离心后再次收集上层提取液,将两次提取液合并浓缩,并经由5 mL/min的二氯甲烷注入GPC,收集馏分后在正己烷中浓缩至1 mL,通过活化二氧化硅柱净化,得到包括BDE-209在内的HFRs总回收率在87.5%~140%,方法检出限范围为0.03~0.65 ng/g。
3.3 人体样品
如图1(c)所示,对于血清等生物体液,液液萃取结合GPC、固相萃取等是最常见的前处理方式。由于GPC分离主要基于分子大小,特别适用于复杂基质条件下不同极性多种目标组分的分析,具有净化效果好、对检测仪器污染小的优势,相较因极性差异而使目标物分离的固相萃取应用更广泛。其中,Hovander等[57]开发的血浆中非极性含卤有机物及其酚类代谢物分析的方法被各实验室广泛采用,通过正己烷/甲基叔丁基醚(体积比1∶1)多次萃取血浆样品,用4 mL正己烷将提取物溶解,分别通过氢氧化钾溶液(0.5 mol/L,50%乙醇,2 mL)和正己烷/甲基叔丁基醚(体积比9∶1,4 mL)分离出非极性化合物及酚类代谢物组分,后续的除脂可使用GPC、硅胶/硫酸活化硅胶柱以及浓硫酸等进行处理,得到多数目标物的回收率在70%~90%,该方法适合包括BDE-209、DBDPE、DP、TBBPA/S-DBPE和TTBP-TAZ等在内的大多数HHFRs的分析。此外,Covaci等[58]针对人体血清中PBDEs建立了基于OASISTMHLB(500 mg)SPE及浓硫酸活化硅胶柱净化的方法,采用二氯甲烷洗脱,其中BDE-209回收率为98%。此外,Shi等[59]将冷冻干燥后的母乳样品无水Na2SO4共研磨后,采用正己烷/丙酮(体积比1∶1)索氏萃取20 h,经GPC净化和浓硫酸除脂后,得到DBDPE的回收率为70%~130%,该方法大大减少了前处理过程中所需耗材和分析污染,为人体血清中痕量分析BFRs提供了参考。
4 HHFRs的分析方法
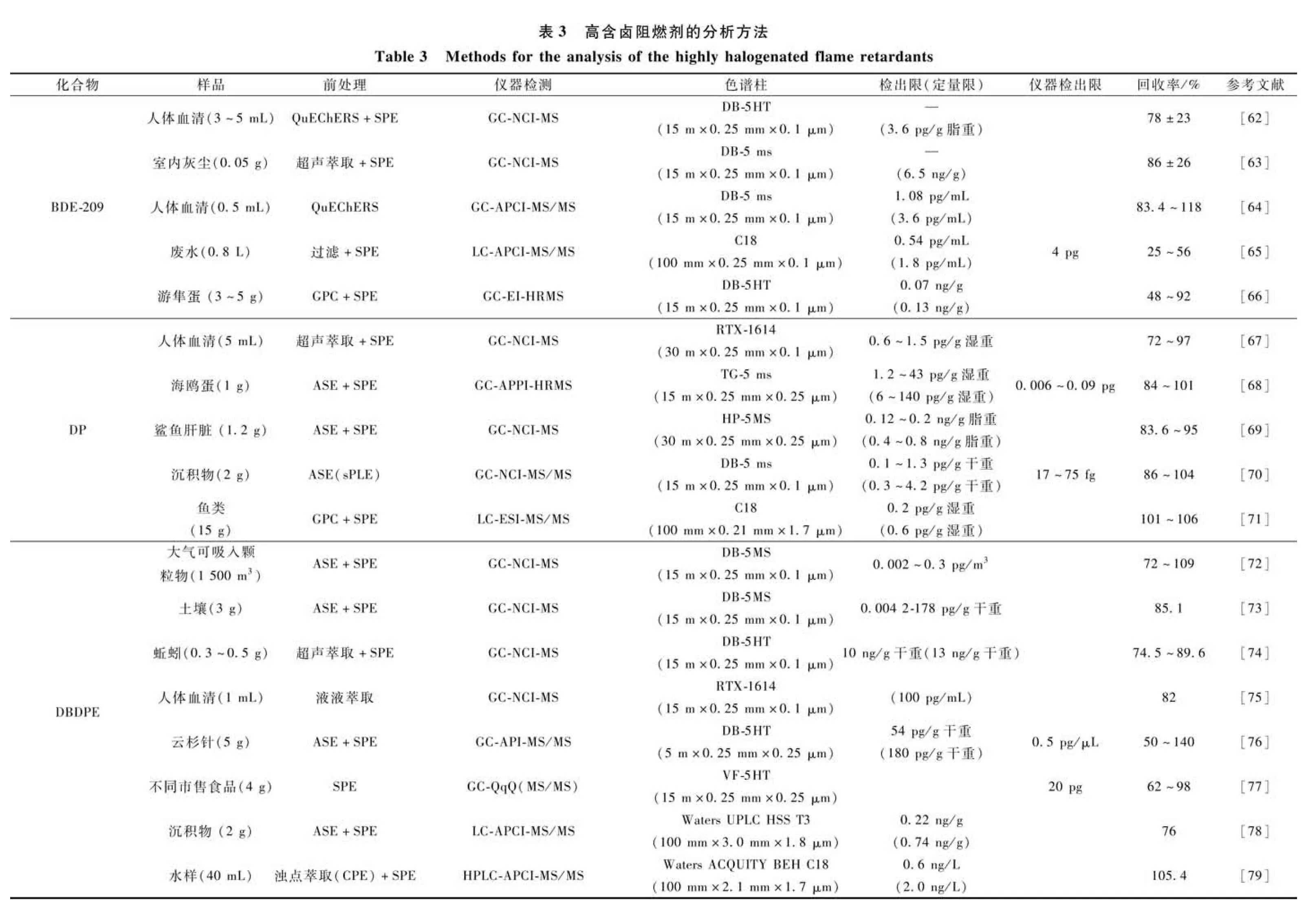
当前HHFRs的仪器检测方法主要是通过气相/液相色谱-质谱法(GC/LC-MS)。GC-MS的电离方式中,相较于常用的电子电离(electron ionization,EI)[60],负化学电离(electron capture negative ionization,NCI)对具有强电负性的HHFRs有高选择性及灵敏度,因而成为分析HHFRs的首选电离方式。NCI有时也被称作电子捕获负电离(electron capture negative ionization,ECNI)。在NCI模式下,目标物的电离方式是电子捕获和反应离子化学电离。电负性分子可以捕获带有很低的动力学能的热电子以产生负离子;反应离子化学电离机制则是分析物与带负电荷的反应气离子反应。由此,NCI属于“软”电离,通过更温和的质子转移过程产生分子离子。在使用NCI 时,由于其高选择性,很多基质或杂质没有响应,从而避免了干扰。目前,大部分文献均采用GC-NCI-MS对HHFRs进行分析,由于HHFRs沸点很高,采用涂层较薄(耐高温)的短色谱柱为更优选择,如15 m×0.25 mm×0.10 μm规格。部分研究也报道了如何克服HHFRs在GC进样口或色谱柱中的降解等难题,如采用程序升温进样口(programmed temperature vaporization,PTV)或者液体直接导入进样口等[61]。
对于LC-MS而言,常用的电离方式有电喷雾电离(electrospray ionization,ESI)、大气压化学电离(atmospheric pressure chemical ionization,APCI)和大气压光电离(atmospheric pressure photoionization,APPI)。由于HHFRs主要是非极性化合物,使用ESI难以电离或电离效率低,通常使用APPI、APCI进行分析。在APCI模式下,样品被喷雾到加热室中,在电晕针帮助下使化合物在气相中离子化;而在APPI模式下,目标分析物在气相中吸收由真空-紫外发出的电子(10 eV或10.6 eV)后放出电子而被离子化。APCI和APPI比较适合非极性或弱极性化合物的分析,不适合热不稳定化合物。表3中列举了各种HHFRs典型的仪器分析方法。
4.1 BDE-209
由于CI源在检测含有强吸电子基团的化合物时,其对负离子的灵敏度远高于其正离子的灵敏度,使得CI源对有机卤素具有高灵敏性和选择性,因此GC/LC与配备CI源的各类质谱联用是环境和生物样品中BDE-209常用的检测方法。Björklund等[84]研究了BDE-209在GC-NCI-MS的质谱碎裂特征,发现BDE-209主要形成醚键断裂的离子簇[C6Br5O]-,m/z(质荷比)范围为482.6~492.6;其次才是Br-的特征峰m/z=79、81。因此,可利用基峰离子m/z=486.6对BDE-209进行定性定量,相比非特异性的Br-的特征离子减小了背景噪声干扰。更重要的是可使用13C12-BDE-209做内标并采用同位素稀释技术进行定量。当13C12-BDE-209作为同位素内标来定量BDE-209时,由于BDE-209与13C-BDE-209有重叠的m/z=488.6、490.6、492.6碎片离子,所以可以分别选择m/z=484.6、486.6作为[12C6Br5O]-的监测离子,494.6和496.6作为[13C6Br5O]-的监测离子。基于该研究,Ma等[62]通过GC-NCI-MS分析了职业暴露人群血清样品中的BDE-209,采用DB-5HT(15 m×0.25 mm×0.1 μm, J&W Scientific, USA)为色谱柱,以m/z=486.7、488.7作为监测离子对BDE-209进行定性定量分析,并采用13C12-BDE-209作为内标进行校正,监测离子为494.7和496.7。并且采用液体直接导入进样口的注入方式及短色谱柱以避免GC系统对BDE-209的降解与吸附,得到的BDE-209定量限为3.6 pg/g脂重。Bu等[63]通过配备DB-5 ms柱(15 m×0.25 mm×0.10 μm, J&W Scientifific, Folsom, CA, USA)的GC-NCI-MS对不同环境室内灰尘样品中的BDE-209进行分析,得到BDE-209的定量限为6.5 ng/g。Wang等[64]开发了气相色谱-大气压化学电离源-串联质谱法(GC-APCI-MS/MS)在MRM模式下分析人体血清样品中的BDE-209。以13C12-BDE-209作为内标,采用DB-5MS柱(15 m×0.25 mm×0.10 μm, J&W Scientifific, Folsom, CA, USA),进样温度为280 ℃,1 μL进样,BDE-209保留时间为10.49 min。由于采用APCI源,能得到大量的准分子离子,因而可以获得比EI更高的信号强度,有助于提高检测的准确度。与GC-NCI-MS和GC-EI-MS/MS相比,新开发的GC-APCI-MS/MS最终得到BDE-209检出限为1.08 pg/mL,远低于GC-NCI-MS法的50 pg/mL和GC-EI-MS/MS法(检出限大于1 ng/mL)。基于气质色谱手段检测BDE-209时,GC进样口高温会导致一定程度的BDE-209降解,而以液质色谱检测可以避免此类问题。Zhou等[65]采用液相色谱-大气压化学电离-三重四级杆串联质谱(LC-APCI-MS/MS)在多反应监测跃迁(MRM)模式下分析废水样品中的BDE-209,以13C12-BDE-209为内标,采用C18柱(100 mm×2.1 mm×2.2 μm, Restek, Bellefonte, PA, USA)进行分析,得到BDE-209的检出限和定量限分别为0.54 pg/mL和1.8 pg/mL。和常规ESI相比,APCI在灵敏度和准确度上具有较显著的优势,适用于多数实验室中HHFRs的分析检测。除了水样,目前通过LC-APCI-MS/MS分析沉积物[78]、食品[85]等样品中的BDE-209应用也较为广泛。
4.2 DP
对DP及其结构类似化合物,如顺式DP(syn-DP)、反式DP(anti-DP)、得克隆602(Dec-602)、得克隆603(Dec-603)以及 DP脱氯产物Cl11-DP、Cl10-DP等,通常采用GC-NCI-MS进行分析测定。Baron等[70]在选择离子监测(SIM)式下使用GC-NCI-MS/MS对沉积物中的DP进行分析测定,以13C-syn-DP作为内标,DB-5MS为色谱柱(15 m×0.25 mm×0.1 μm, J&W Scientifific, Folsom, CA, USA),DP仪器检出限在17~75 fg。在分析中,可选用不同色谱柱以及内标物对多种样品中的DP进行监测,也都呈现出较高的灵敏度和分辨率。例如通过配备RTX-1614柱(30 m×0.25 mm ID×0.1 mm, Restek Inc, USA)的GC-NCI-MS对人体血清中的DP进行分析[67],以13C-syn-DP和13C-anti-DP为内标(监测离子为m/z=661.7、663.7),设置syn-DP和anti-DP的监测离子为m/z=651.7、653.7,得到DP的检出限为0.6~1.5 pg/g脂重。分析检测鲨鱼肝脏样品中DP及其类似物[69]时,以3′-氟-2,2′,4,4′,5,6′-六溴二苯醚(F-BDE-154)作内标,HP-5MS为色谱柱(30 m×0.25 mm×0.25 μm, J&W Scientific, Agilent),得到DP的定量限为0.4~0.8 ng/g脂重。此研究中设置离子源温度为150 ℃,用以避免离子的高度碎裂。虽然基于GC-NCI-MS的方法有较高的特异性及灵敏度,但较低的离子源温度使离子源受污染而频繁清洗,导致仪器工作效率下降。由而具有高分辨率的高分辨质谱(HRMS)可以精确测定化合物的质荷比,上述研究通过GC-APCI-HRMS,还鉴定出与Dec-603结构相关的两种新型脱氯烷类化合物,为DP类化合物的鉴定提供了新的途径。Ayala-Cabrera等[68]采用气相色谱-大气压光电离-高分辨质谱(GC-APPI-HRMS)来测定海鸥蛋样品中DP及其相关化合物,以13C12-CB-209作为内标,采用TG-5MS色谱柱(15 m×0.25 mm×0.25 μm, Thermo Fisher Scientific, San Jose, CA, USA),设置进样温度为280 ℃,离子源温度为250 ℃,乙醚作为溶剂有机掺杂剂,得到DPs的检出限和定量限分别为1.2~43 pg/g脂重、6~140 pg/g脂重,避免了低温下离子源的反复清洁。
4.3 DBDPE
由于DBDPE与BDE-209结构相似,主要差别仅在于苯环间的碳-碳键与醚键,因此针对DBDPE的仪器分析方法与BDE-209类似。Zhang等[72]开发了一种通过GC-NCI-MS检测大气可吸入颗粒物中DBDPE的分析方法,采用DB-5MS(15 m×0.25 mm×0.1 μm, J&W Scientifific, USA)为色谱柱,设置程序升温程序以及进样口、传输线、离子源温度(分别为280、300、250 ℃),以氦气为载气(流速1.2 mL/min),得到DBDPE的检出限为0.002~0.3 pg/m3。此外,Xiong等[73]基于此方法,对土壤样品中的DBDPE进行检测,得到DBDPE的检出限为0.004 2~178 pg/g干重,证明该仪器分析方法针对不同环境样品中的DBDPE分析具有良好的适用性。Klimm等[16]通过类比DBDPE和十溴联苯(PBB-209)在紫外光作用下的脱溴特征,在GC-NCI-MS的基础上,以DB-5HT(15 m×0.25 mm×0.1 μm, Agilent, Waldbronn, Germany)为色谱柱,通过监测Br-1的特征峰m/z=79、81以及两个高分子量的碎片离子[M-Br]-(m/z=884.3监测DBDPE)、[M-Br+H]-(m/z=805.1监测九溴BDPEs,m/z=726.9监测八溴BDPEs),结合GC-EI-MS的质谱碎片特征,推断并识别了3个八溴BDPEs(BDPE-197,BDPE-201和BDPE-202)和3个九溴BDPEs(BDPE-206,BDPE-207和BDPE-208),此外,通过GC-NCI-Orbitrap HRMS验证了DBDPE含氧转化产物(OxyTPs)的存在。和BDE-209一样,DBDPE也会由于GC进样口高温发生降解,因此如何解决此问题是研究关注的要点。Neugebauer等[76]在采用气相色谱-大气压电离-三重四级杆串联质谱(GC-API-MS/MS)对云杉针中的DBDPE进行定量分析时,以13C-DBDPE作内标,DB-5HT(15 m×0.25 mm×0.25 μm, Agilent, USA)为色谱柱,分别设置进样衬管和传输线温度为280 ℃和340 ℃,尽可能提高仪器分析速度,减少DBDPE在系统中的停留时间,避免DBDPE降解。同时,在分析过程中设置梯度流量,显著提高仪器检出性能,得到DBDPE的仪器检测限和方法定量限分别为0.5 pg/μL和180 pg/g干重。此外,Garcia-Bermejo等[77]建立了针对市售食品中DBDPE含量的气相色谱-三重四级杆串联质谱(GC-QqQ-MS/MS)检测方法,在MRM模式下以m/z=485为前体离子,m/z=325、404分别为定量离子和定性离子监测DBDPE,采用VF-5HT色谱柱(15 m×0.25 mm×0.25 μm, Lake Forest, CA, USA),调控并优化MS操作参数、电离能量和灯丝发射电流,使分子离子特别是溴化度高的离子碎片减少,最大限度地提高目标化合物的响应水平,最终在40 eV和50 μA的组合条件下得到的DBDPE的仪器检出限为10 pg。由于DBDPE在GC高温程序下的热不稳定性,研究者一直致力于开发LC-MS检测分析DBDPE的方法,目前针对含DBDPE样品分析检测的LC-MS手段也具有良好的灵敏性,如Feng等[78]通过配备Waters UPLC HSS T3色谱柱(100 mm×3.0 mm×1.8 μm, Waters, Milford, USA)的LC-APCI-MS/MS对沉积物样品中DBDPE进行检测,在0.25 mL/min流速条件下采用线性梯度洗脱,得到的DBDPE检出限和定量限分别为0.22 ng/g和0.74 ng/g。Zhou等[79]开发了一种HPLC-APCI-MS/MS分析水样中DBDPE的方法,以Waters ACQUITY BEH C18(100 mm×2.1 mm×1.7 μm)为色谱柱,通过对流动相溶剂类型和比例进行优化,最终确定在流动相A、B分别为0.02 mol/L乙酸铵水溶液和甲醇/乙腈(体积比90∶10),流动相流速0.3 mL/min,柱温40 ℃条件下获得最优的色谱分离和灵敏度,得到DBDPE的检出限和定量限分别为0.6 ng/L和2.0 ng/L,并且通过同位素内标13C14-DBDPE的校正提高了该方法的可重复性。由于高溴化合物在GC高温程序下的热不稳定性导致有时很难确保诸如DBDPE分析的准确性和灵敏度,通过溴片段进行定量也使得NCI难以区分经GC色谱柱共洗脱的含溴HHFRs,而采用配备APCI的LC-MS手段则很好地解决了这一问题,并且由于其操作简便和高灵敏度而成为DBDPE环境分析检测中的主流方法之一。
4.4 TBBPA/S-DBPE
GC-MS和高效液相色谱-质谱法(HPLC-MS)是常用的检测 TBBPA/S-DBPE的方法。然而,GC-MS直接分析易导致TBBPA/S-DBPE热分解,如Ali[51]等在开发针对TBBPA-DBPE等新型溴化阻燃剂的检测方法中,由于GC-NCI-MS中衬管和DB-5 ms色谱柱温度条件的影响造成目标物降解使回收率(48%)并不理想,因此GC-MS一般不是 TBBPA/S-DBPE的首选方法[86]。对于HPLC-MS,通常会引入ESI、APCI和APPI等多种离子源来满足复杂样品的分析需求,但它们在监测TBBPA/S-DBPE的应用中受到一定的限制,部分原因是仪器和化合物的特性造成电离困难。例如,Qu等[87]在对HPLC-ESI-MS/MS和HPLC-APCI-MS/MS的方法优化中,发现在ESI模式的全扫描优化下没有产生任何显著的TBBPA-DBPE前体离子,而APCI模式下虽然可获得TBBPA-DBPE的特异性光谱,但其灵敏度不足以检测TBBPA-DBPE在实际环境样品中的浓度。Letcher等[80]开发了一种通过LC-APPI-MS/MS在APPI负模式下检测TBBPA/S-BDPE的分析方法,其中LC配备ACE 3 C18分析柱(2.1 mm×50 mm×3 μm)和ACE 3 C18保护柱(2.1 mm×10 mm×3 μm, Aberdeen, UK),流动相为水和丙酮,流速为0.2 mL/min,流动相梯度如下:0%丙酮相于进样后0.01 min升至100%丙酮相,保持15 min,再于1 min升至100%水相,保持14 min。该梯度下降低了背景噪声,从而提高了信噪比,使得TBBPA/S-BDPE色质谱均获得了良好的峰形。通过MRM模式实现对TBBPA/S-BDPE的高灵敏度定量,其中TBBPA-DBPE的监测离子为m/z=975.5、79,TBBPS-DBPE的监测离子为m/z=997.4、79,得到的TBBPA-BDPE方法检出限和定量限分别为0.07 ng/g脂重和0.25 ng/g脂重,仪器检出限为32 pg;TBBPS-DBPE方法检出限和定量限分别为1.28 ng/g脂重和4.27 ng/g脂重,仪器检出限为112 pg。APPI虽然对TBBPA/S-BDBPE检测灵敏且具有特异性,但需要额外相匹配的掺杂剂用于电离[88],难以适应大多数分析实验室的日常需求。因此针对TBBPA/S-DBPE仪器分析技术的开发也面临巨大挑战。
目前,基于高效液相色谱-二极管阵列检测器(HPLC-DAD)立体化检测TBBPA/S-DBPE得到开发和应用,尤其在定性和定量方面发挥极大优势。Liu等[19]采用HPLC-DAD和HPLC-MS/MS串联ESI源分别检测螺类和鱼类等海洋生物中的TBBPA-DBPE、TBBPS-DBPE,以D10标记的TBBPA(10 ng)作为内标,ZORBAX ODS为色谱柱(150 mm×3 mm×5 μm, Agilent),得到的TBBPA-DBPE检出限和定量限分别为6 ng/g干重和20 ng/g干重,仪器检出限为3 ng;TBBPS-DBPE检出限和定量限分别为20 pg/g干重和60 pg/g干重,仪器检出限为1 pg,这也是首次报道在生物样品中检测到TBBPS-DBPE。随后该课题组首次建立了一种基于薄层色谱(TLC)样品预处理和HPLC-DAD(UV=214 nm)相结合的新方法[54],可用于测定含TBBPA-DBPE和TBBPS-DBPE的土壤样品。选用Zorbax ODS柱(150 mm×3 mm×5 μm, Agilent)为色谱柱,以乙腈和水(含0.1%甲酸)为流动相,流速为0.6 mL/min时,分离效果最好,且TLC具有更高的样品通量、更短的分析时间和更少的提取和清理步骤,为后续HPLC-DAD的检测有效的消除了干扰,得到TBBPA-DBPE和TBBPS-DBPE的检出限分别为36 ng/g和49 ng/g,定量限分别为120 ng/g和160 ng/g,该法快速且经济高效。除此之外,HPLC-DAD联合其他技术在环境及生物样品中检测TBBPA/S-DBPE得到广泛应用[89-90],也成为目前检测TBBPA/S 及其衍生物的重要手段之一。同时,对于TBBPA-DBPE转化产物,可采用超高分辨率傅里叶变换离子回旋共振质谱(FTICR-MS)进行初步筛选[37],利用超高性能液相色谱-轨道阱高分辨率质谱(UHPLC-Orbitrap HRMS)对污染区域土壤样品进行定性和定量分析,以13C12标记的TBBPA作为内标,得到的目标转化产物检出限为0.02~3 ng/g干重。该方法具有高灵敏度和良好的可重复性,可用于其他复杂的生物和环境基质中TBBPA/S-DBPE的分析。
4.5 TTBP-TAZ
在已有研究报道中,气质色谱和液质色谱均可对TTBP-TAZ进行分析检测。Guo等[31]采用GC-EI-HRMS对灰尘中TTBP-TAZ进行分析时,采用RTX-1614为色谱柱(15 m×0.25 mm×0.1 μm, Restek Corporation, Bellefonte, CA),以BDE-118为内标,得到TTBP-TAZ的检出限为0.6 ng/g。作者通过EI-HRMS扫描样品时,根据TTBP-TAZ结构分析,该化合物可以失去一个溴原子形成以m/z=987为中心的离子簇,或失去其三个溴化环(R=C6H2Br3)中的一个形成以m/z=754 为中心的离子簇。但对于TTBP-TAZ这种低挥发性高分子量化合物,在使用GC-MS测定时,对样品的前处理要求高,且出现结果不稳定的现象。因此,目前液质是测定TTBP-TAZ的主流方法。Baesteros-Gomez等[29]开发了一种基于直接探针与大气压化学电离-高分辨率飞行时间-质谱(APCI-TOF-HRMS)结合的方法,通过直接探针可将固体样品直接引入 MS 源,省去样品制备和色谱分离步骤,可以对环境样品进行快速筛查;同时,采用溶剂萃取法,通过LC-APCI-TOF-HRMS确认筛选结果,并定量测定塑料和粉尘样品中TTBP-TAZ的水平,得到TTBP-TAZ的检出限和定量限分别为20 ng/g和60 ng/g。这个检测方法也在后续有关WEEE污染的聚合物玩具材料中TTBP-TAZ口腔迁移研究中得到了应用[91],此外,ESI离子源对TBBP-TAZ进行测定可以得到较好的电离效果。Shen等[30]采用HPLC-ESI-MS/MS在MRM模式下检测电子垃圾回收区的灰尘样品,TTBP-TAZ检出限和定量限分别3.8 ng/g干重和12.7 ng/g干重。MRM 模式选择性和抗干扰能力强,灵敏性高,可直接将样品过滤后上机测试,无需再用有机溶剂提取转移。而LC-MS/MS 可通过灵活调节离子化模式来测定TTBP-TAZ,如Lorchner等[21]采用LC-ESI-MS/MS对沉积物中的TBBP-TAZ进行了分析,以13C18-TTBP-TAZ作内标,选用C18为色谱柱(100 mm×2 mm×3 μm, Phenomenex©, Aschaffenburg, Germany),总运行时间为35 min。在使用该方法时,需要对色谱方法进行优化,选择水(0.1%甲酸)和甲醇/乙腈(80∶20)溶液为两种流动相,设置柱温40 ℃、流速0.25 mL/min时可得到最好的峰形和分离结果,TTBP-TAZ检出限和定量限分别为10.2 ng/g和27.8 ng/g。这样可避免严重的基质干扰,极大提高灵敏度。
4.6 TTBNPP
作为一种新型HHFRs,目前关于TTBNPP相关研究报道较少。在采用气质联用方法进行测定时,不同离子源模式下的检测效果差异较大。Halloum等[92]发现,在EI正模式的全扫描MS中,未检测到TTBNPP的分子离子,TTBNPP在m/z=145处检测到碎片离子,推断为[C5H6Br]+,具有特异性的峰为丢失一个侧链的m/z=712.6;而在APCI正模式的MS中可检测到TTBNPP的[M+H]+离子以及失去一个溴的[M—Br]+离子。在两种离子源串联GC-MS/MS对加标鱼类样品的检测中,EI未得到任何TTBNPP可供分析的结果,而APCI检测TTBNPP得到的相对标准偏差值为43%,说明该检测方法无法获得TTBNPP的准确定量结果。而采用液质联用方法能够对环境样品中的TTBNPP进行定性和定量分析。McGoldrick等[81]采用配备C18柱(100 mm×2.1 mm×3.5 μm)的LC-ESI-MS/MS在正离子模式下对鱼样中的TTBNPP进行检测,虽然并未在实际环境鱼样中检测到TTBNPP,但通过加标实验得到的TTBNPP检出限与定量限分别为0.16 ng/g脂重和0.49 ng/g脂重,为检测TTBNPP提供了一种良好可重复的方法。Santin等[82]采用LC与配备ESI源的四极杆线性离子阱串联质谱(QqLIT-MS/MS)结合的方法在SRM模式下对鱼类样品中的TTBNPP进行了分析,色谱柱采用RP-18柱(125 mm×2.0 mm×5 μm, Merck, Darmstadt, Germany),流动相为水(0.1%甲酸)和甲醇(10 mM醋酸铵),最终得到TTBNPP的检出限和定量限分别为37.4 ng/g脂重和125 ng/g脂重,仪器检出限和仪器定量限分别为58.8 pg和196 pg。Lu等[83]开发了一种超高效液相色谱-大气压化学电离-串联质谱(UPLC-APCI-MS/MS)在APCI正离子模式下检测海鸥蛋中TTBNPP的方法,以C18柱(50 mm×2.1 mm×1.6 μm, Phenomenex, CA, USA)为色谱柱,在MRM模式下通过监测离子m/z=1 018.14、226.89、144.96对TTBNPP进行定性定量,得到TTBNPP的检出限和定量限分别为0.02 ng/g脂重和0.1 ng/g脂重,显著低于LC-ESI-MS/MS。该方法通过使用UPLC使得待测组分获得更高的分离度和灵敏度,与LC相比在含TTBNPP样品的痕量分析中更具优势。
5 结论与展望
已有研究表明,HHFRs产用量逐年增加,在各类环境介质中普遍存在(图2),势必会对生态环境和人群健康带来较大的潜在威胁。但目前国内外对其环境行为归趋和风险的研究尚未全面展开,仍存在以下问题。
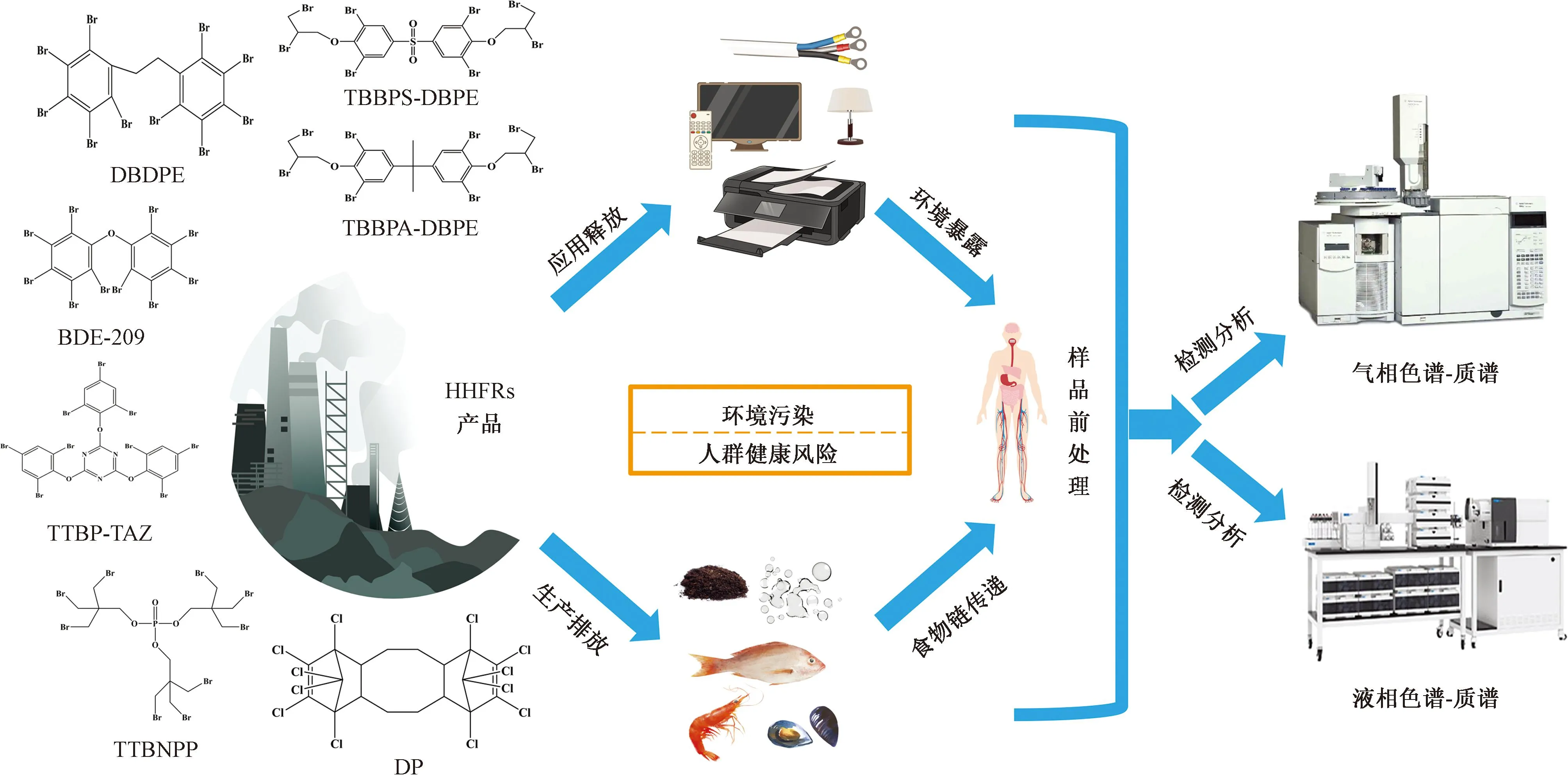
图2 HHFRs研究概括图Fig.2 Research summary of HHFRs
(1)已报道的HHFRs数据主要围绕北美(美国、加拿大)、欧洲和东亚(中国、日本、韩国)的少数国家,而主要电子产品的进口国如南美、俄罗斯、印度等地以及许多发展中国家的HHFRs数据报道相当有限;BDE-209、DBDPE和DP等传统的HHFRs受到关注较多,是环境介质及人体内检出率及浓度较高的HHFRs,但有关TBBPA/S-DBPE、TBBP-TAZ、TTBNPP的研究有待进一步展开。
(2)对于前处理技术,传统HFRs的SPE、液液萃取等方法也适用于HHFRs的提取与净化,通过对萃取溶剂和吸附填料的选择和优化能实现较好的萃取率。但HHFRs和一般HFRs在物化性质上存在较大差异,传统前处理技术对多种HHFRs在萃取效率、样品通量以及复杂基质条件下的萃取还存在较大缺陷。
(3)对于仪器分析,当前主流的分析手段是将HHFRs与其他HFRs通过气质或液质联用进行同步分析,这样较单一分析方法更节省时间和成本。但由于HHFRs的特质以及主流仪器分析手段存在的优缺点,如何实现同步分析和表征复杂样品中可能存在的全部HHFRs及其转化产物对研究者来说是很大的挑战。
(4)在气质联用中,NCI对大多数HHFRs的敏感性使其成为目前质谱中常用的电离首选。然而由于多数含溴HHFRs在配备NCI的MS中主要通过产生溴片段作为信号用于定量,因此NCI不能区分与GC色谱柱共洗脱的含溴HHFRs,MS检测器中选择性的缺乏导致想要在GC色谱柱中更好地分离HHFRs的难度增大。
(5)对液质联用而言,APPI和APCI是分析HHFRs的主要电离技术,而APPI对紫外灯和掺杂剂的要求使其在大多数分析实验室中的适用性相当有限。如何基于APCI的液质手段,实现在单份复杂基质样品中,将待测HHFRs目标组分有效分离出峰将是下一步方法开发中的巨大挑战。
考虑到HHFRs与一般HFRs性质的差异,如HHFRs的高度疏水性、低挥发性和难电离等特点,并结合目前的研究现状和存在的问题,今后相关的研究应集中在下面几个方面。
(1)结合ASE、QuEChERS等高效的新型前处理方法,开发提取时间短、溶剂消耗少、分离净化步骤简单的复杂基质中多种HHFRs前处理技术;建立质控体系,提高不同实验室研究数据间的可比对性。
(2)对于分析方法,既要开发适用于精准定性定量分析的常规方法,通过优化色谱质谱分析检测条件、色谱柱和离子源等参数以提升分析方法的灵敏度和结果的可靠性;又要结合先进手段如直接探针(DP)方法与APCI-TOF-HRMS相结合的快速筛选方法,对环境介质中的HHFRs污染源进行快速识别。
(3)系统开展HHFRs转化产物的分析,亟需合成更多的HHFRs转化产物单体以及相应的同位素内标,并且能够通过商品化途径获取,以便国内外学者针对HHFRs的环境归趋及毒性效应等方面开展更深入的研究,为准确评估其潜在生态和健康风险提供科学依据。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
色谱(2022年11期)2022-11-10
色谱(2022年10期)2022-10-13
色谱(2022年7期)2022-06-25
波谱学杂志(2022年2期)2022-06-14
色谱(2022年4期)2022-04-01
世界科学技术-中医药现代化(2020年2期)2020-07-25
中成药(2018年12期)2018-12-29
中成药(2017年6期)2017-06-13
医学研究杂志(2015年4期)2015-06-10
