
基于共沉积法的高结晶反蛋白石g-C3N4制备及其产氢性能分析
2024-03-12 09:29颉启东傅雅琴司银松
浙江理工大学学报(自然科学版) 2024年1期
颉启东,王 聪,傅雅琴,司银松
(浙江理工大学材料科学与工程学院,杭州 310018)
0 引 言
21世纪以来,随着煤炭、石油和天然气等石化能源的过度消耗,能源短缺和环境污染问题愈发明显[1-2]。能源利用从不可再生的石化能源向可再生能源的过渡已成为能源领域的研究热点[3-4]。半导体光催化技术利用光催化剂将太阳能转变为氢能,氢能具有较高的能量密度和燃烧热值,在燃烧过程中燃烧产物是水,没有二氧化碳等污染性气体产生,可以实现循环利用,为解决能源危机提供了良好的途径[5-6]。然而,目前半导体光催化剂存在太阳光利用率低、载流子复合率高等问题,导致催化剂的活性较低,限制了光催化剂的实际应用[7]。因此,开发高活性的半导体光催化剂至关重要[8]。
石墨相氮化碳(g-C3N4)是一种新兴的半导体光催化剂,具有物理化学性质稳定、成本低和无毒无污染等优势,在半导体光催化领域具有广阔的应用前景[9-11]。但由含氮前驱体直接高温煅烧获得的块状g-C3N4通常存在晶粒尺寸较大、比表面积较小等问题,限制了光催化反应过程中活性位点的利用和载流子的迁移[12-13]。同时,这类催化剂在高温热聚合过程中易于产生晶格缺陷,严重阻碍了光生电子-空穴分离,进一步降低了催化剂的活性[14-15]。
通过同时提高比表面积和结晶程度可以有效增加g-C3N4光催化活性[8,12]。反蛋白石多孔结构因具备独特的形貌特征,拥有该类结构的光催化剂不仅可以提高比表面积,增加光催化反应活性位点,而且还可以将入射光多重散射,加强对太阳光的有效利用,被广泛用于提高g-C3N4光催化活性的制备中[16-17]。目前,国内外已经报道了反蛋白石结构g-C3N4在光催化领域的应用。王有和等[18]以离心自组装制备的蛋白石结构SiO2胶体为硬模板剂,单氰胺为前驱体制备出反蛋白石多孔结构g-C3N4材料;Sun等[19]通过无裂纹、高度有序的胶体模板制备出反蛋白石结构g-C3N4;Lin等[20]通过使用两步纳米铸造的方法合成3 D有序紧密堆积g-C3N4纳米微球阵列。然而,上述文献制备的反蛋白石结构g-C3N4仍存在结晶程度较低和制备工艺流程复杂的问题,限制了反蛋白石结构g-C3N4光催化性能的提升。因此,开发出一种简单、有效的合成方法,制备出均匀稳定的高结晶反蛋白石结构g-C3N4,对获得性能优异的半导体光催化剂具有重要意义。
本文采用共沉积法,通过二氧化硅微球/单氰胺混合溶液制备不同结晶程度的反蛋白石g-C3N4,通过扫描电镜、X射线衍射仪、红外光谱仪和比表面积及孔径分析仪等表征手段对其结构和形貌进行分析;在模拟太阳光照射下采用气相色谱仪对其光催化产氢性能进行分析,并探究了结晶程度对反蛋白石结构g-C3N4的光催化产氢性能的影响。
1 实验部分
1.1 实验材料与仪器
1.1.1 实验材料
正硅酸乙酯(AR)和三乙醇胺(AR)购自上海阿拉丁生化科技有限公司,单氰胺(95%)和氟化氢铵(99.5%)购自上海麦克林生化科技有限公司,单氰胺水溶液(质量分数50%)购自西格玛奥德里奇(上海)贸易有限公司,氯铂酸水溶液(1.34 mg/mL)购自南京化学试剂有限公司,无水乙醇(AR)购自杭州高晶精细化工有限公司,超纯水(H2O,R=18.2 MΩ·cm)由Plus-E2超纯水机(南京易普易达科技发展有限公司)提供。
1.1.2 实验仪器
电子天平(YP1201N,上海精密科学仪器有限公司);电热鼓风干燥箱(DHG-9030A,上海精宏实验设备有限公司);管式炉(OTF-1200X-S-DVD,合肥科晶材料技术有限公司);高速离心机(TG1650-WS,上海沪粤明科学仪器有限公司);磁力搅拌器(MYP11-2,上海梅颖浦仪器仪表制造有限公司)。
1.2 实验方法
1.2.1 SiO2微球的制备
SiO2微球的制备参考文献[21],具体方法如下:将98 mL超纯水、51 g无水乙醇和36 g氨水混合均匀后,获得溶液A;接着将143 g乙醇和18 mL正硅酸乙酯混合均匀后配成溶液B,在室温搅拌条件下,然后将溶液B加入溶液A中,继续反应24 h。再将离心、洗涤后的样品置于50 ℃的烘箱中干燥,制备出SiO2微球。
1.2.2 反蛋白石结构g-C3N4的制备
首先称取1.0 g的SiO2微球,超声分散至3.5 g的氰胺溶液(质量分数50%)中,将所得溶液放入离心机中,通过使用不同的离心速度控制SiO2微球的重力沉积效果,离心时间为3 min,离心速率如表1所示,制备出不同蛋白石结构的SiO2@NH2CN胶体沉积液。接着倒出上层清液,将沉积物在50 ℃烘箱中干燥。然后将干燥后的混合物在管式炉中以2 ℃/min的升温速率升温到550 ℃,保温4 h后,自然冷却到室温,得到SiO2@g-C3N4。最终通过氟化氢铵溶液(4 mol/L)对SiO2@g-C3N4进行刻蚀48 h除去SiO2,制备出不同结构的g-C3N4(记为CNx,其中x表示为离心速度)。作为对照,使用单氰胺为前驱体,以2 ℃/min的升温速率升温到550 ℃,保持4 h,制得块状g-C3N4,记为BCN。
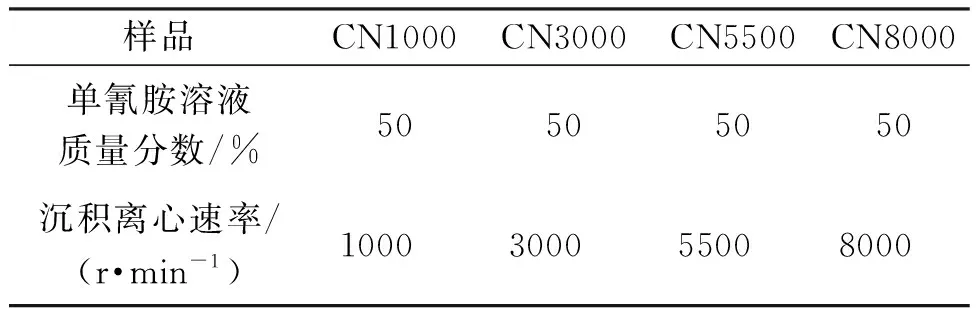
表1 反蛋白石结构g-C3N4的不同沉积离心速率
1.2.3 高结晶反蛋白石结构g-C3N4的制备
高结晶反蛋白石结构g-C3N4的制备流程如图1所示。首先将1.0 g的SiO2微球均匀分散至3.5 g的氰胺水溶液(质量分数50%)中,然后加入不同质量的单氰胺(95%)提高单氰胺溶液质量分数,将溶液放入离心机中,均使用5500 r/min的共沉积离心速度,离心时间为3 min,制备出不同浓度的蛋白石结构的SiO2@NH2CN胶体沉积物。接着将其在50 ℃烘箱中干燥,然后在管式炉中以2 ℃/min的升温速率升温到550 ℃后保持4 h,自然冷却至室温,煅烧出SiO2@g-C3N4。最后使用氟化氢铵溶液(4 mol/L)对SiO2@g-C3N4溶液胶体进行刻蚀48 h除去SiO2模板,制备出不同结晶程度的反蛋白石结构g-C3N4(记为CN5500-y,其中y表示溶液中单氰胺的质量分数),所用单氰胺溶液质量分数如表2所示。

图1 高结晶反蛋白石结构g-C3N4的制备流程示意图

表2 高结晶反蛋白石结构g-C3N4的不同单氰胺溶液质量分数
1.3 测试与表征
使用X射线衍射仪(XRD,D8 discover,Bruker,德国)分析样品的晶体结构,扫描速率为2(°)/min,扫描范围为10°~80°;通过场发射扫描电子显微镜(FESEM,ZEISS Sigma 300,日本)观察样品的表面形貌特征;使用X射线光电子能谱仪(XPS,Thermo Scientific K-Alpha+,美国)分析样品的元素和化学价态;通过红外光谱仪(FT-IR,Nicolet 5700,美国)检测样品官能团,使用KBr压片法进行制样,仪器波长扫描范围为4000~500 cm-1;使用稳态/寿命荧光光谱仪(FLS1000,Edinburgh Instruments,英国)表征样品载流子复合程度;使用比表面积及孔径分析仪(BET,ASAP2020HD88,美国)分析样品的比表面积和孔径分布;使用紫外分光光度计(UH-4150,Hitachi Corporation,日本)表征试样的带隙和光吸收能力。
光催化产氢测试:在光催化制氢系统(CEL-SPH2N,Ceaulight,中国)中评估光催化活性。将20 mg样品置入40.0 mL超纯水中,接着加入10.0 mL三乙醇胺和0.445 mL氯铂酸溶液制,得分散溶液;超声波处理15 min后,将溶液保持在真空环境,使用300 W氙灯(Ceaulight CEL-HXF300,中国)照射反应溶液,进行光催化产氢反应。在整个反应期间使用冷凝系统保持反应溶液温度为6 ℃,并使用高纯氮气作为载体,通过气相色谱仪(GC D7860,TCD Detector,5 Å Molecular Sieve Column,中国)分析所得氢气产量。
2 结果与讨论
2.1 形貌分析
图2为不同离心速率(1000~8000 r/min)所制备样品的SEM图。从图2中可以看出,不同离心速率所制备的样品均出现多孔结构。当离心速率为5500 r/min时,反蛋白石结构g-C3N4(CN5500)的大孔均匀排列且规整有序,大孔尺寸约为290 nm,与SiO2微球的粒径一致;大孔中还会形成1~3个小孔,尺寸约20 nm。小孔可以作为大孔相互贯通的通道,不仅有利于光催化反应过程中太阳光更好地进入,形成多次反射和散射的效果,而且有利于液体反应介质和反应产物的快速传质[19],进而提升产氢速率。

图2 反蛋白石结构g-C3N4的SEM图
图3为CN5500的低倍率SEM图。图3显示,通过共沉积法制备出反蛋白石结构g-C3N4的大孔均匀且排列规整,证明该制备方法能够稳定一致地获得反蛋白石结构g-C3N4[18]。

图3 CN5500的低倍率SEM图
2.2 比表面积和孔结构分析
不同离心速率(1000~8000 r/min)制备出样品的比表面积和孔径分布采用N2吸附-脱附测试表征,结果如图4和图5所示。从图4(a)和图5(a)可以看出,随二氧化硅微球/单氰胺沉积离心速率的增加,比表面积表现出先增大后减小的趋势。在离心速率为5500 r/min时,CN5500具有最大的比表面积(32 m2/g),是块状g-C3N4(5 m2/g)的6倍多,所有样品都表现出Ⅳ等温线和H4型滞后环,证明了在不同条件下获得的样品均具有纳米多孔结构[12,22]。从图4(b)和图5(b)可以看出:样品的孔径分布集中在5 nm和20 nm附近,证明样品具有介孔结构,与SEM表征结果一致;当保持离心速率为5500 r/min时,当只增加溶液中的单氰胺溶液质量分数时,样品的比表面积反而逐渐降低,孔径分布趋于不明显,其原因可能是更多前驱体的存在,在热聚合过程中形成了块状g-C3N4,导致比表面积的减小。
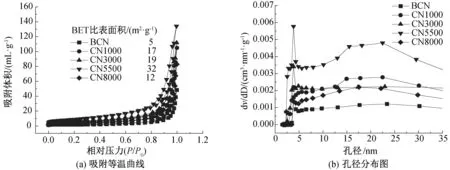
图4 反蛋白石结构g-C3N4的N2吸附-脱附曲线及孔径分析图

图5 高结晶反蛋白石结构g-C3N4的N2吸附-脱附曲线及孔径分析图
2.3 晶体结构和微观结构分析
通过XRD来表征反蛋白石结构g-C3N4的结晶性能,结果如图6所示。从图6中可以看出,所有样品在2θ为13.1°和27.4°处皆出现g-C3N4的2个特征衍射峰,其中位于13.1°的(100)峰对应于g-C3N4平面内三嗪结构单元的规整排列,而位于27.4°的(002)峰对应于π共轭平面的层间堆叠[10,23],表明成功制备了g-C3N4。在不同离心速度下,相同单氰胺溶液质量分数(50%)条件下制备的CNx(002)峰的衍射角均位于27.3°且无明显偏移,说明在不同转速下合成的g-C3N4与块状g-C3N4均具备相似的晶体结构(图6(a))。当增加单氰胺溶液质量分数至70%时,CN5500-70%(002)峰的2θ增加至27.56°,与CN5500相比明显向右偏移;CN5500-70%的层间距从3.27 Å减小到3.23 Å(图6(b)),表明了CN5500-70%的π共轭平面层间堆叠更加致密[12,24]。g-C3N4的电子传输主要作用在垂直于平面结构之间的通道中,较短的层间距离可以提升光催化过程中的电子迁移速率,因此可以提高g-C3N4的光催化活性[25-26],表明在反蛋白石结构g-C3N4的聚合过程中,通过增加单氰胺溶液质量分数,可以产生更多的中间体产物,使其聚合更加完整,从而提高了反蛋白石结构g-C3N4的结晶程度[26-27]。
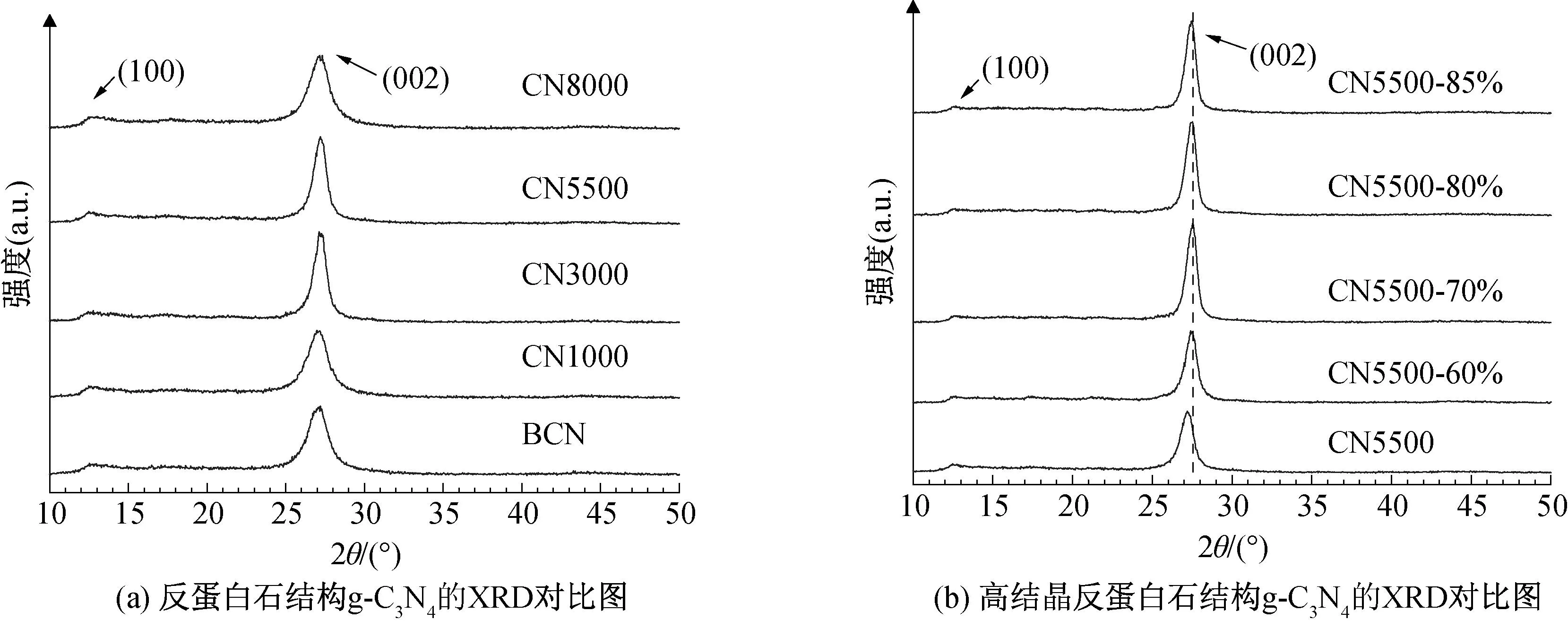
图6 不同结晶程度反蛋白石结构g-C3N4的XRD谱图
通过图7对比图2(c)可以发现,高结晶反蛋白石结构g-C3N4提高了结晶程度,同时还能继续保持良好的反蛋白石多孔结构,表明一定范围内提高单氰胺溶液质量分数不会破坏反蛋白石结构的形成。
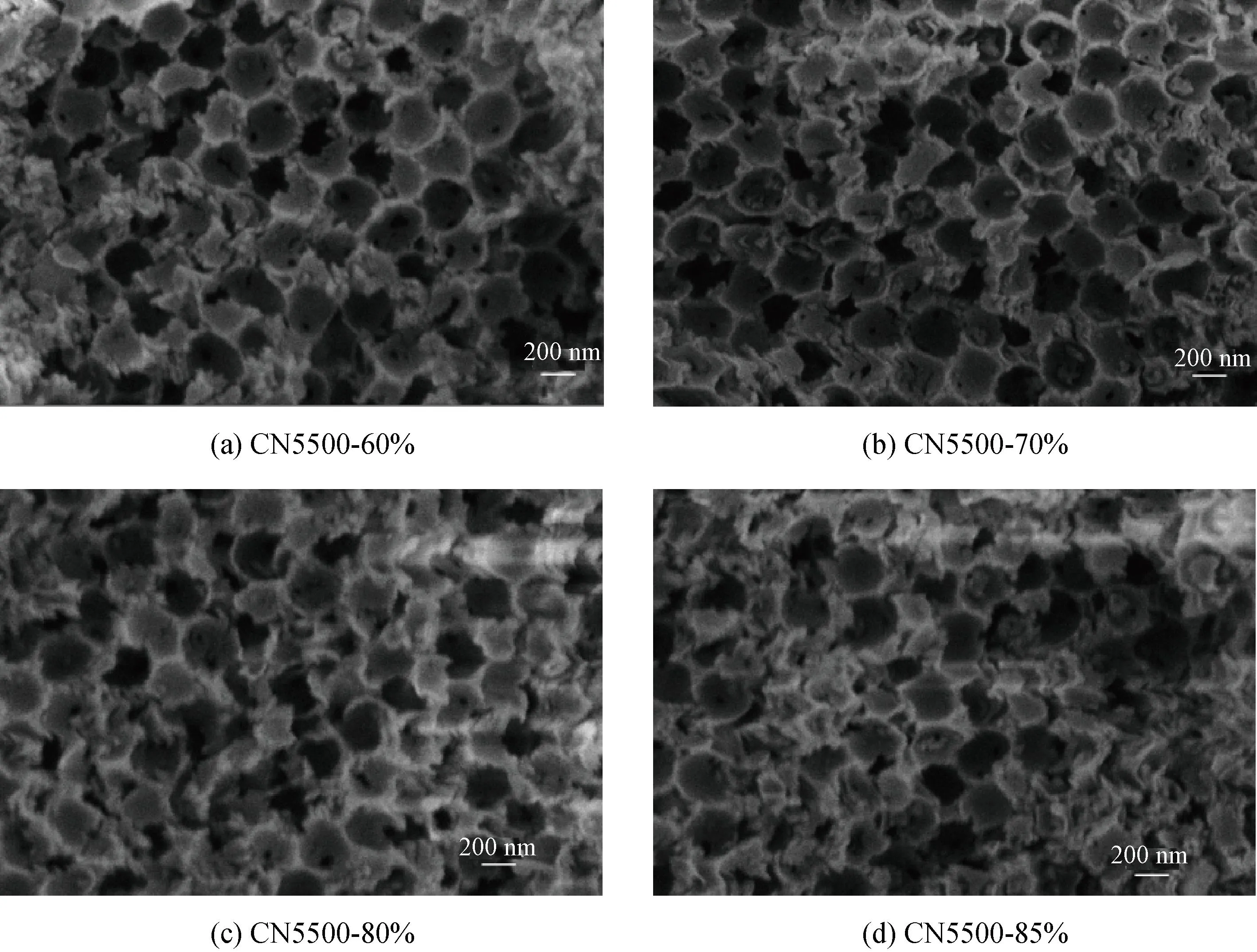
图7 高结晶反蛋白石结构g-C3N4的SEM图
2.4 内部官能团和表面原子状态
通过FT-IR对样品中的官能团进行表征,结果如图8所示。从图8(a)可以看出,所有样品的FT-IR谱图的峰型均与块状g-C3N4的FT-IR谱图相似,表明所制备样品的官能团种类并没有产生明显变化,位于810 cm-1的吸收峰为三嗪环的伸缩振动,在1200~1600 cm-1吸收带是C—N键与—HN(C2)的伸缩振动,位于3000~3500 cm-1范围的峰是残余氨基基团(—NHx)引起的伸缩振动,或者是样品表面吸附的水分子的伸缩振动[12,28];随着溶液中单氰胺质量分数的提高,所有反蛋白结构样品位于810 cm-1与1200~1600 cm-1附近的吸收峰均有所增强,是样品聚合程度和结晶性能的提高所致[28];所有样品在2253 cm-1左右的位置出现一个较小的峰,是由于氰基(—C≡N)的不对称拉伸振动引起的,表明样品仍具有氰基缺陷,这可能会限制光催化活性[12]。CN5500-70%位于此处峰面积的减小显示氰基缺陷被修复,形成了更加致密的聚合物网络结构,可能有利于光催化反应的进行,提高g-C3N4的光催化活性。

图8 高结晶反蛋白石结构g-C3N4的FT-IR、XPS谱图

2.5 光吸收分析
图9为高结晶反蛋白石结构g-C3N4的UV-vis光谱。与块状g-C3N4相比,样品的吸收带向蓝光方向大幅度移动(见图9(a)),表明高的结晶程度增大了g-C3N4的带隙;从图9(b)可以看出,高结晶反蛋白石结构g-C3N4的带隙从2.52 eV(CN5500)增加至2.62 eV(CN5500-70%),并随着单氰胺溶液质量分数的增加,带隙逐渐减小至2.60 eV(CN5500-85%)。

图9 高结晶反蛋白石结构g-C3N4的UV-Vis光谱
图10为高结晶反蛋白石结构g-C3N4的能带图。从图10中可以看出,高结晶反蛋白石结构g-C3N4的带隙先增大后减小,带隙变化是因为结晶程度的不同导致价带(VB)能量最高点位置和导带(CB)能量最低点位置发生变化[31],CN5500-70%的导带位置是所有样品中最小的,导带位置越负,说明光生电子对H+具有更强的还原性,有利于光催化反应过程中H2的产生[12,31];随着单氰胺溶液质量分数的增加,带隙减小至2.60 eV(CN5500-85%),是因为CN5500-85%的导带位置相比于CN5500-70%向下移动,使光生电子对H+的还原反应能力变弱,可能会导致CN5500-85%光催化活性的降低[30,32]。
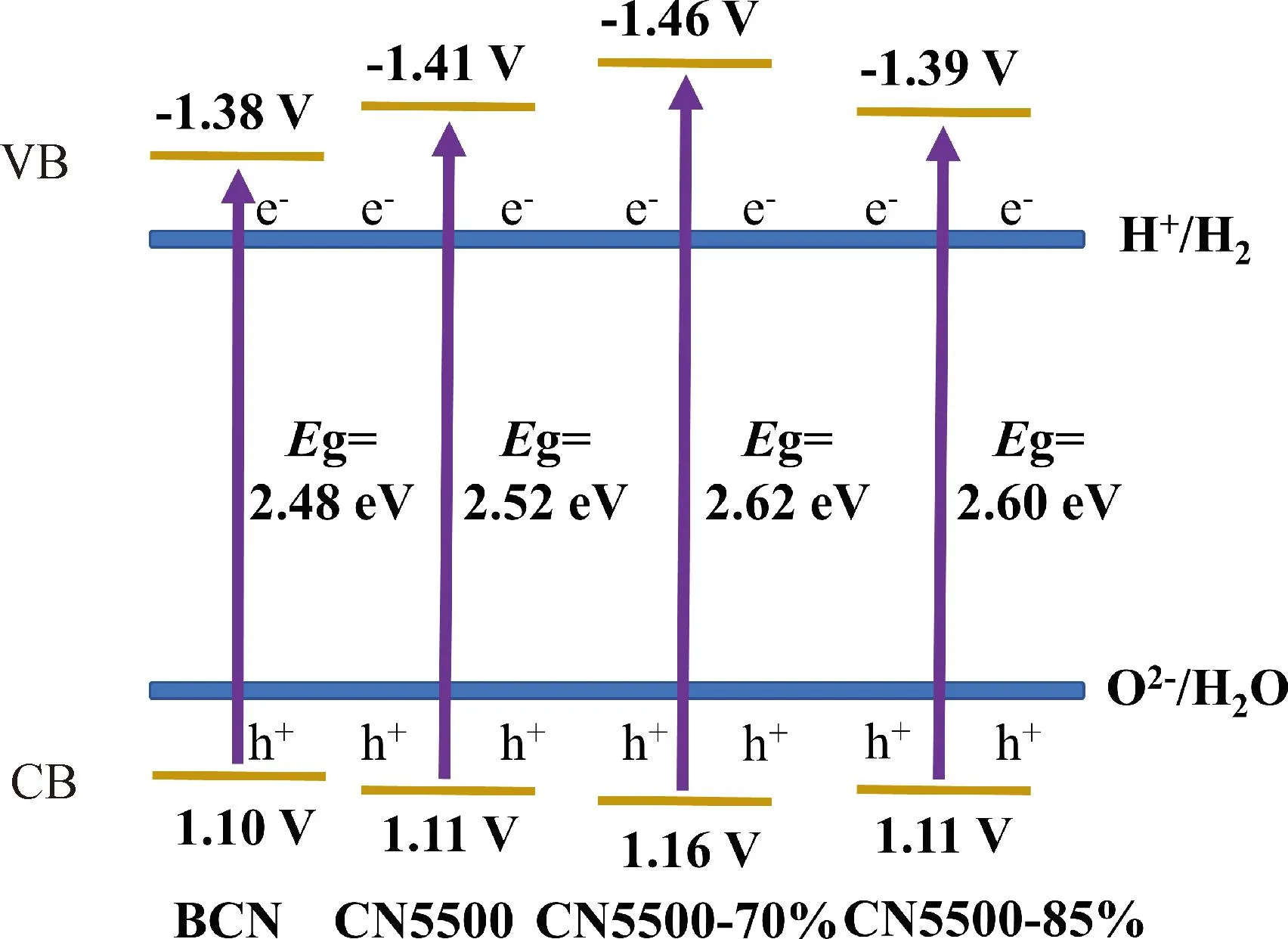
图10 高结晶反蛋白石结构g-C3N4的能带图
在光催化反应过程中,载流子的分离与迁移也是影响光催化活性的重要因素。通过使用稳态荧光光谱(PL)来表征光生电荷载流子的分离和转移过程。从图11可以看出,随单氰胺的质量分数提高,PL强度最高时的峰位置从CN5500的450 nm向左偏移至CN5500-70%的430 nm,随后又向右偏移至CN5500-80%的470 nm,说明较高的结晶程度可以提高对可见光的吸收[31],与UV-vis图谱表现一致;高结晶反蛋白石结构g-C3N4比BCN的位于最高点位置的PL强度要低得多,说明提高结晶程度可以有效减少载流子的复合[12,33],高结晶反蛋白石结构g-C3N4的PL强度随质量分数的增加表现为先减小后增大,在CN5500-70%处降到最低,表明一定质量分数下(70%)合成的高结晶反蛋白石结构g-C3N4可以有效抑制光生电子-空穴的复合[29,33],这是因为结晶程度的增强导致了内部缺陷的减少,与本文之前讨论的FT-IR表征结果一致。
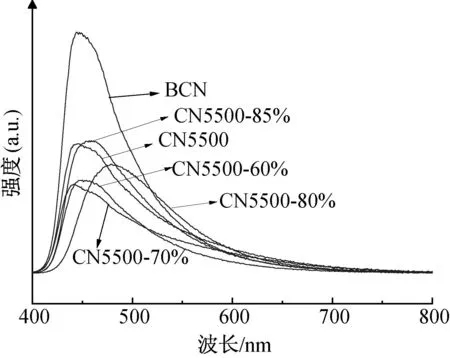
图11 高结晶反蛋白石结构g-C3N4的PL图
2.6 光催化产氢性能
通过使用Pt(质量分数3%)和三乙醇胺(TEOA)作为助催化剂和牺牲剂,对g-C3N4的光催化产氢性能进行了分析,结果如图12所示。图12表明:受SiO2胶体沉积速度的影响,样品的产氢速率随沉积速率的增加表现出先增大后减小的趋势,CN5500的光催化性能表现最佳,产氢速率为4137.19 μmol/(g·h);在同一沉积速率(5500 r/min)下,提高单氰胺质量分数时,样品产氢速率随质量分数的增加表现为先增大后减小,CN5500-70%的产氢性能最好,产氢速率高达7217.01 μmol/(g·h),表明增强的结晶程度结合反蛋白石多孔结构显著提高了g-C3N4的光催化产氢活性。

图12 不同结晶程度反蛋白石结构g-C3N4的产氢速率
高结晶反蛋白石结构g-C3N4的光催化产氢性能提高的机理可以从以下方面解释:首先,当光线照射到高结晶反蛋白石结构g-C3N4材料上时,由于其独特的表面结构,使太阳光在大孔中进行多重散射,部分光线可以通过贯穿小孔进入更深的内部,增加了太阳光的有效利用,这是使反蛋白石结构g-C3N4(CN5500-y)产氢速率普遍提高的原因;其次,CN5500的比表面积比块状g-C3N4的比表面积大约6倍,可以提供更多的活性位点,因而提高了其光催化反应活性,使产氢速率提升至4137.19 μmol/(g·h);最后,当反应溶液中单氰胺的质量分数为70%时,有效提高了反蛋白石结构g-C3N4的结晶程度,从而提升了g-C3N4层与层之间的电荷迁移速率,有利于光催化水解过程中对于H+的还原,从本质上增强了反蛋白石结构g-C3N4的产氢速率,CN5500-70%在模拟太阳光照射反应下表现出最佳的光催化产氢性能,产氢速率高达7217.01 μmol/(g·h),是块状g-C3N4产氢速率(1209.25 μmol/(g·h)的6倍。
3 结 论
本文采用共沉积法制备出高结晶反蛋白石结构g-C3N4,并探究了结晶程度的提升对g-C3N4光催化产氢性能的影响。主要研究结果如下:
a)在不同的沉积速率下制备的g-C3N4的比表面积均有所提升。CN5500呈现出典型的反蛋白结构,大孔均匀且排列规整有序,比表面积增大至32 m2/g,有利于提供更多的活性位点,使产氢速率提升至4137.19 μmol/(g·h)。
b)通过控制单氰胺溶液质量分数在70%时,可以有效增加反蛋白石结构g-C3N4的结晶程度。CN5500-70%的层间距缩短至3.23 Å,光生电子和空穴复合速度明显降低,在模拟太阳光照射反应下表现出最佳的光催化产氢性能,产氢速率高达7217.01 μmol/(g·h),是块状g-C3N4产氢速率(1209.25 μmol/(g·h))的6倍,证明了规整有序的三维多孔结构和高结晶对g-C3N4光催化活性提升的协同作用。
本文采用的共沉积法简单有效,所制备的高结晶反蛋白石结构g-C3N4既保留了三维有序的反蛋白石结构,又增强了g-C3N4的结晶程度,显著增强了反蛋白石结构g-C3N4光催化产氢性能。
猜你喜欢
学与玩(2022年12期)2023-01-11
陶瓷学报(2019年5期)2019-01-12
无机盐工业(2017年5期)2017-05-25
郑州大学学报(理学版)(2017年1期)2017-04-07
三峡大学学报(自然科学版)(2017年1期)2017-03-20
化工管理(2017年25期)2017-03-05
中国塑料(2016年9期)2016-06-13
中国资源综合利用(2016年9期)2016-01-22
中国塑料(2015年7期)2015-10-14
应用化工(2014年7期)2014-08-09
