
XPO1基因突变的慢性淋巴细胞白血病临床特征及预后研究
2024-03-13 04:46韩宜霏许张娣吴佳竹孔祎琳潘必慧梁金花申浩睿李建勇
南京医科大学学报(自然科学版) 2024年3期
韩宜霏,许张娣,吴佳竹,孔祎琳,潘必慧,李 悦,梁金花,申浩睿,尹 华,王 莉,李建勇,徐 卫
南京医科大学第一附属医院血液科,江苏 南京 210029
慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)是一种慢性B 淋巴细胞克隆增殖性疾病,表现为CD5+的成熟B淋巴细胞在外周血、骨髓、淋巴结和脾脏中聚集,具有显著的遗传和临床异质性[1]。近年来,随着分子靶向治疗的飞速发展,如布鲁顿酪氨酸蛋白激酶(Bruton tyrosine kinase,BTK)抑制剂、磷酸肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)抑制剂、BCL-2 抑制剂等,极大地改善了患者的预后,CLL 的治疗由传统免疫化疗时代逐步进入无化疗时代[2],但仍有部分患者会出现治疗耐药,预后相对较差。而大规模的测序研究包括二代测序(next-generation sequencing,NGS)越来越多地应用于血液系统肿瘤研究,极大程度上增加了入们对CLL 基因组改变的认识,也逐步揭示了由克隆异质性导致的临床异质性[3]。
目前CLL中多个再现突变的驱动基因已被报道,包括TP53、NOTCH1、SF3B1、BIRC3、MYD88、ATM、XPO1、FBXW7 和POT1 等,并确定克隆演变是驱动疾病进展的重要机制[4-6]。XPO1基因突变存在于多种血液系统肿瘤中,尤其是B细胞淋巴瘤和白血病,包括原发纵隔大B 细胞淋巴瘤(primary mediastinal large B-cell lymphoma,PMBCL)、经典型霍奇金淋巴瘤(classical Hodgkin lymphoma,cHL)、弥漫大B细胞淋巴瘤(diffuse large B-cell lymphoma,DLBCL)和CLL[7]。在西方国家,XPO1 基因在CLL 中的突变频率为2.4%~8%[8-9],低于侵袭性B 细胞淋巴瘤。XPO1 基因位于2 号染色体长臂(2p15),其编码的XPO1 蛋白是重要的核输出蛋白,介导含有核输出信号(nuclear export-signal,NES)结构的蛋白由细胞核向细胞质的转运,对维持细胞稳态是必不可少的[10-11]。已知XPO1 常见的热点突变为E571K 或E571G,处于NES 结构域相邻位置[7]。多项研究表明XPO1 E571K 突变可能促进B 细胞淋巴瘤的发生发展[12-13]。鉴于XPO1 基因突变在CLL 致病及发生Richter 综合征过程中具有重要地位,本研究对本中心携带XPO1 基因突变患者的资料进行回顾性分析,增加对我国携带XPO1基因突变的CLL的认识,以期为临床诊治提供帮助。
1 对象和方法
1.1 对象
本回顾性研究纳入了2006 年11 月—2022 年3 月于南京医科大学第一附属医院血液科确诊CLL的543 例患者。纳入标准:年龄≥18 岁;符合《中国慢性淋巴细胞白血病/小淋巴细胞淋巴瘤的诊断与治疗指南(2018 年版)》[14]的诊断标准;同意参加本研究者。排除标准:样本资料严重缺失。所有患者均进行外周血/骨髓形态检测、骨髓病理免疫组织化学染色、流式细胞术免疫分型、荧光原位杂交(fluorescence in situ hybridization,FISH)、免疫球蛋白(immunoglobulin,IG)重链基因重排和CpG+白细胞介素-2(interleukin-2,IL-2)刺激的染色体核型分析等检测。同时,采用PCR法检测免疫球蛋白重链可变区基因(immunoglobulin heavy-chain variable region,IGHV)突变状态和NGS 检测相关基因突变状态。CLL 诊断标准、分期、CLL 国际预后指数(CLL-international prognostic index,CLL-IPI)评分和疗效评估均严格参照《中国慢性淋巴细胞白血病/小淋巴细胞淋巴瘤的诊断与治疗指南(2018年版)》[14]。本研究已获得南京医科大学第一附属医院伦理委员会审核批准(伦理号:2023-SRFA-272)。所有患者均知情同意。
1.2 方法
1.2.1 XPO1基因检测方法
①样本类型:所有患者均在获得知情同意后抽取新鲜外周血或骨髓液标本。②检测方法:提取单个核细胞,进行分选。采用PCR扩增法检测。③检测深度:1500*。④变异等位基因频率(variation allele frequency,VAF)计算:VAF(%)=突变reads/(突变reads+野生reads)×100%。
1.2.2 随访及预后
本研究随访截止时间为2022 年6 月。随访方式主要为门诊及住院病历查阅和电话咨询。至首次治疗时间(time to first treatment,TTFT)定义为自明确诊断到开始接受治疗的时间;无进展生存时间(progression-free survival,PFS)定义为自首次治疗到疾病进展的时间或末次随访时间;总生存时间(overall survival,OS)定义为自明确诊断到任何原因导致死亡或随访终点的时间。
1.3 统计学方法
采用SPSS 27.0软件进行统计学分析,计数资料用频数和构成比[n(%)]表示。计量资料用中位数(四分位数)[M(P25,P75)]表示。计量资料的组间比较采用t检验分析。生存曲线由Kaplan-Meier 方法构建,各组患者生存曲线比较使用Log-rank 检验。Graphpad Prism 9.4 软件进行生存曲线绘图。P<0.05表示差异有统计学意义。
2 结果
2.1 病例特征
在543例CLL患者中,根据NGS检测时状态,分为初诊未治(treatment native,TN)患者368例,复发/难治(relapsed/refractory,R/R)患者175 例。15 例(2.8%)患者XPO1基因突变检测阳性,TN、R/R患者的突变率分别为2.4%(9/368)及3.4%(6/175),其中1例患者TN和R/R时均进行了NGS检测,且均检测出XPO1 基因突变。在15 例XPO1 突变患者中,男性占66.7%(10/15),女性占33.3%(5/15)。TN和R/R患者的临床特征见表1。结果显示,无论TN 还是R/R 患者,男性比例均多于女性(TN:55.6%vs.44.4%;R/R:83.3%vs.16.7%),且疾病大多处于RaiⅢ/Ⅳ期,Binet B/C 组。TN 组中位外周血白细胞绝对计数为24.5(8.9,52.7)×109个/L、淋巴细胞绝对计数为17.4(5.6,44.0)×109个/L、血红蛋白为86(76,116)g/L、血小板计数为191(118,200)×109个/L,中位β2-微球蛋白(β2-microglobulin,β2-MG)为3.0(2.9,4.2)mg/L和乳酸脱氢酶(lactate dehydrogenase,LDH)为294(198,329)U/L;R/R组中位外周血白细胞计数为28.4(20.2,56.2)×109个/L、淋巴细胞计数为24.1(13.2,52.0)×109个/L、血红蛋白110(91,118)g/L、血小板计数为92(60,139)×109个/L,中位β2-MG 为4.8(3.5,7.6)mg/L和LDH为256(165,388)U/L。
2.2 XPO1突变位点及变异等位基因频率(variation allele frequency,VAF)分析
15 例CLL 患者携带XPO1 突变基因,均为错义突变,且存在热点突变,中位VAF 为35.4%。66.7%的XPO1基因突变发生在外显子15的E571位点,分别为E571K(50%)、E571G(30%)、E571Q(10%)和E571V(10%)(图1A、B)。同时,本研究发现,R/R CLL组相较于TN CLL组,XPO1的VAF略高,但两者差异并无统计学意义(图1C)。此外,在CLL患者中XPO1突变并不是一个孤立的克隆性突变,XPO1突变常伴随TP53、KMT2D、SF3B1、NOTCH1 和ATM 等基因突变同时发生。图1D~H 展示了在XPO1 突变伴随TP53、KMT2D、SF3B1、NOTCH1 和ATM 基因突变同时发生的患者中,XPO1 与伴随突变基因VAF的比较。鉴于XPO1 基因编码的XPO1 蛋白具有重要的核输出功能,而p53 蛋白是其转运的重要核输出蛋白之一,因此,本研究进一步比较了XPO1基因突变与TP53 基因突变同时出现时的患者临床特征。结果表明,TP53 与XPO1 基因突变同时发生多见于R/R 组(TN:11.1%;R/R:50.0%),在TP53 与XPO1基因突变同时发生的患者中,XPO1的VAF高于TP53 的VAF,但两者差异并无统计学意义(图1D)。1 例患者在R/R 时XPO1 VAF 水平相比TN 时升高,且出现了新的基因突变(ATM、KMT2D 和DDX3X突变)(图1I)。
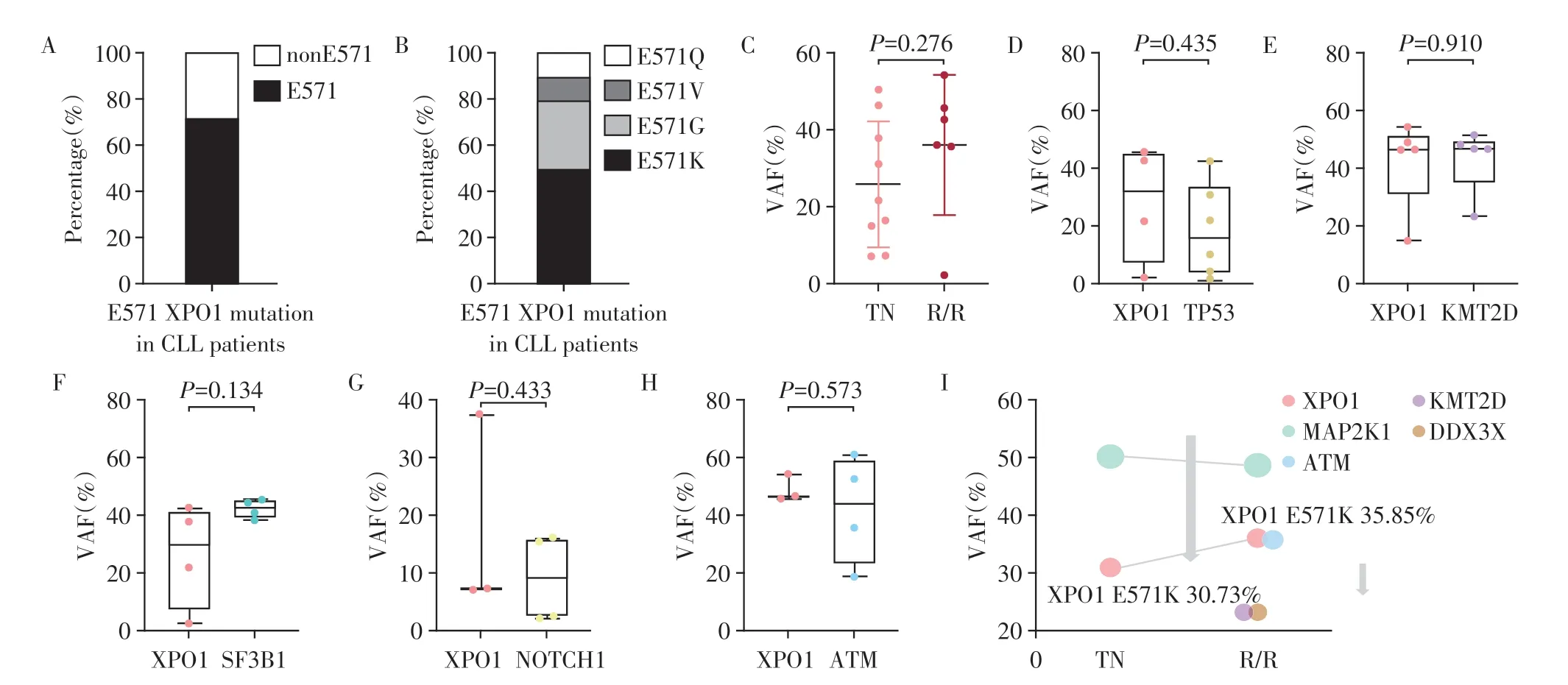
图1 XPO1基因突变位点和基因突变的VAF Figure 1 XPO1 mutation locus and VAF of gene mutations
2.3 XPO1突变患者的分子和遗传学特征
在15 例患者中,11 例(73.3%)患者IGHV 无突变,4 例(26.7%)患者IGHV 有突变,4 例(26.7%)患者染色体为复杂核型(≥3 个异常染色体)。发生XPO1 基因突变患者的分子和遗传学特征见表2。在TN 组中,XPO1 基因突变常伴随NOTCH1、SF3B1、KMT2D 和ARID1A 基因突变发生,25.0%的患者FISH 可见ATM 缺失、37.5%的患者可见13q 缺失,并无患者存在17p 缺失。而在R/R 组中,XPO1基因突变常伴随TP53、KMT2D 和ATM 基因突变发生,且60.0%的患者FISH 可见ATM 缺失和13q 缺失,40.0%的患者可见17p 缺失。TN 组中位突变数目为4,R/R组中位突变数目为5(图2)。
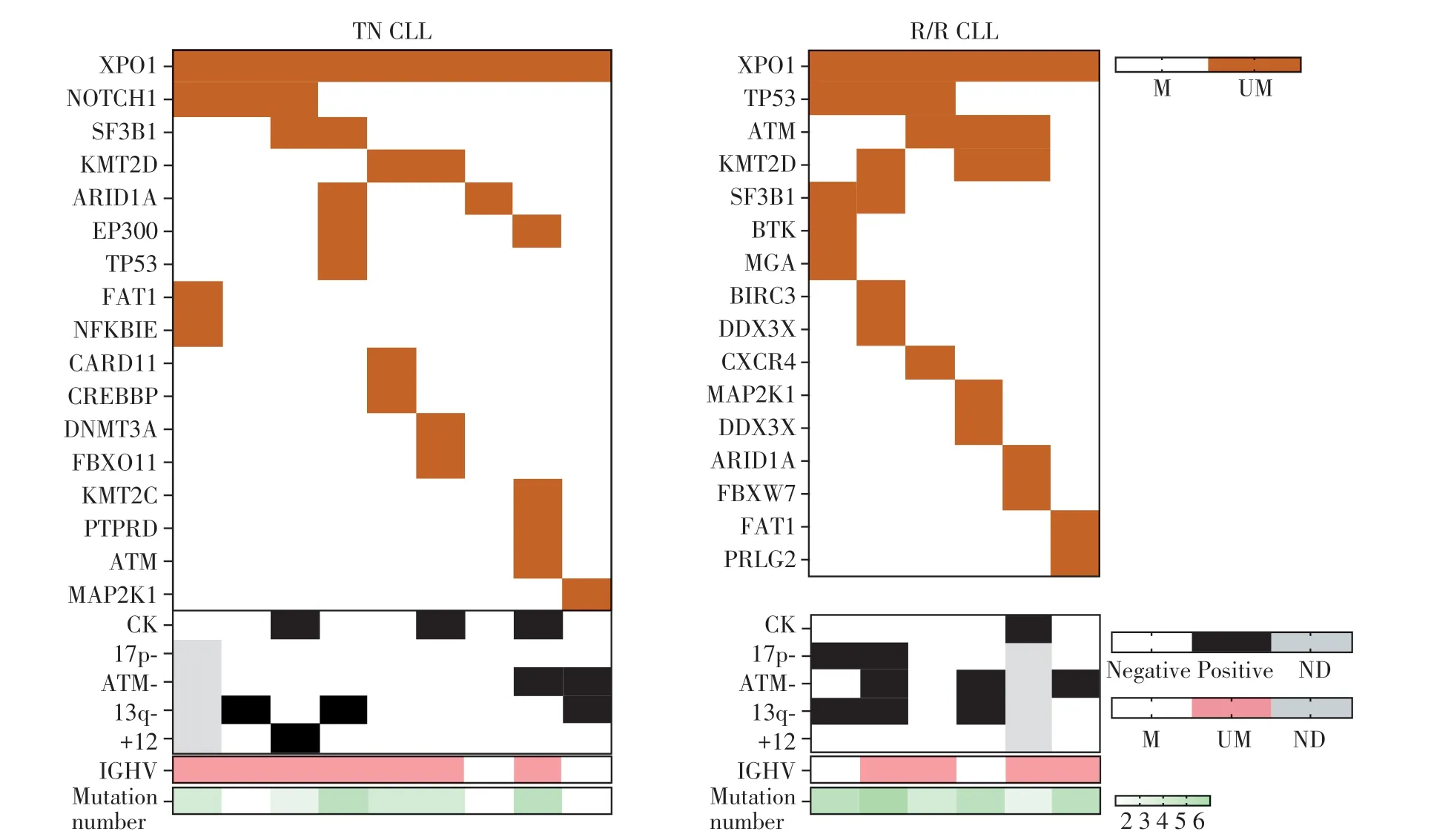
图2 15例携带XPO1突变的CLL患者队列中基因突变、分子和遗传学特征分布Figure 2 Distribution of gene mutations,molecular and genetic characteristics in the cohort of 15 CLL patients with XPO1 mutations

表2 15例XPO1突变CLL患者遗传学和分子特征Table 2 Genetic and molecular characteristics of 15 CLL patients with XPO1 mutations
2.4 XPO1突变对治疗及预后的影响
在15 例XPO1 突变患者中,除3 例未达到治疗指征,其余12例已经接受治疗。对于9例TN患者,3例患者目前处于观察等待期,3例患者已接受治疗并获得部分缓解(partial response,PR),2 例患者治疗后获得完全缓解(complete response,CR),1 例患者治疗后转为难治。而对于6 例R/R 患者,在疾病进展后,均进行BTK抑制剂(伊布替尼或泽布替尼)治疗,4例患者获得缓解;2例患者疾病控制不佳,其中1例患者在泽布替尼治疗耐药后出现中枢受累,另1 例患者则发生了Richter 综合征,均已死亡(图3A)。截至2022年6月,XPO1突变组患者中位TTFT为1.8个月,中位PFS为19.8个月,中位OS为40.0个月;XPO1 无突变组患者中位TTFT 为8.1 个月,中位PFS 为32.5 个月,中位OS 为49.8 个月。两者TTFT(P=0.633)、PFS(P=0.394)和OS(P=0.799)差异均无统计学意义(图3B~D)。TN 时XPO1 突变组和无突变组采用BTK 抑制剂化疗的总反应率(overall response rate,ORR)为100.0%vs.80.5%(P=0.651)。R/R 时XPO1 突变组和无突变组采用BTK 抑制剂化疗的ORR 为66.7%vs.56.0%(P=0.927)。另外,26.7%(4/15)的患者发生了Richter综合征。
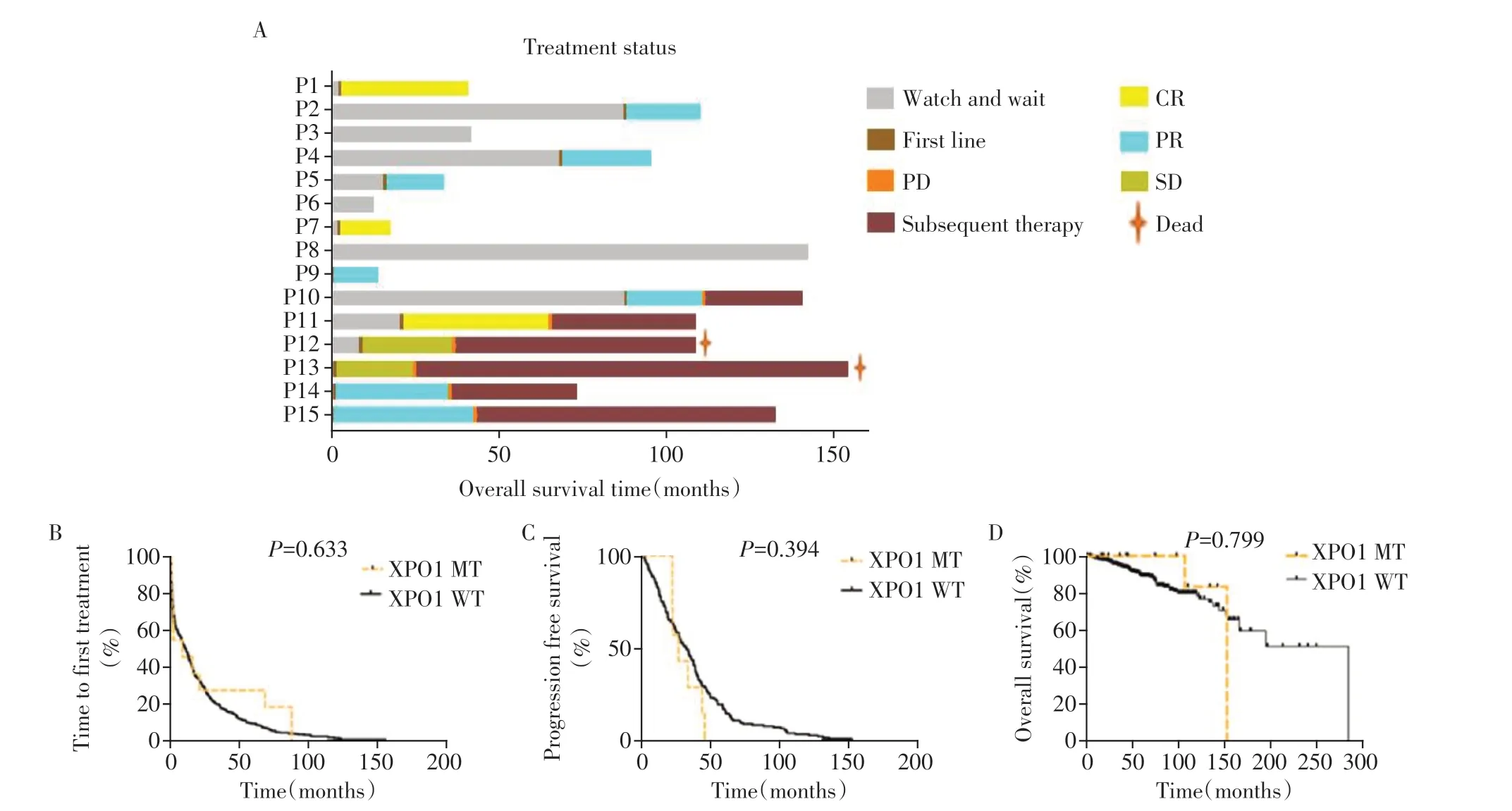
图3 15例携带XPO1突变的CLL患者治疗情况、疗效评估、生存结局和预后等特征分布Figure 3 Distribution of treatment status,efficacy evaluation,survival outcome and prognosis of 15 CLL patients with XPO1 mutations
3 讨论
XPO1 基因突变广泛存在于各种实体瘤和血液系统肿瘤中,而重现性E571位点突变多发生在B细胞恶性肿瘤中。CLL是一种常见的B淋巴细胞克隆增殖性疾病。2011 年,Puente 等[9]通过全外显子测序首次揭露了重现性XPO1基因突变在CLL中的致病作用。目前国外已有针对XPO1 基因突变与CLL患者临床特征的相关性研究,但国内尚缺乏相关报道。国外研究显示,XPO1 基因突变频率为2.4%~8%[8-9]。在本中心数据中,XPO1突变也是一个低频突变,突变率低于3%,略低于国外。此外,XPO1基因突变均为错义突变,且中位VAF为35.4%。
目前越来越多的突变复杂性/肿瘤突变负荷(tumor mutation burden,TMB)评估将XPO1突变纳入其中,通过评估TMB来预测CLL患者预后。如2018年Nadeu 等[15]将28 个CLL 相关驱动基因测序,并说明TMB 与CLL 患者较短的无治疗生存(treatment-free survival,TFS)相关。2021年Chauzeix等[16]研究显示ATM、SF3B1、NOTCH1、XPO1、MYD88、TNFAIP3 和TP53组成的基因模型可以较好预测较短的TFS,甚至对Binet A期的患者也能准确预测。由此可见XPO1基因突变在CLL患者预后评估中具有重要意义。
在CLL 患者中,XPO1 突变与高危预后指标相关。2015 年西班牙的一项2 493 例CLL 队列中,XPO1突变和TP53突变一样,存在于ATM缺失患者中,该类患者预后不佳[17]。Puente等[9]也发现XPO1突变主要存在于IGHV无突变的CLL患者。本研究结果与之一致,绝大多数XPO1 基因突变患者为IGHV无突变,其TTFT相比IGHV有突变患者更短,且60%的R/R患者存在ATM缺失。本研究还发现,R/R 患者一线治疗采用传统免疫化疗,但均未获得持续缓解。这部分患者末次治疗选用BTK抑制剂,2/3 患者获得缓解,未获得缓解的2 例患者中1 例同时伴随TP53 和BTK(C481S)突变。而TN 患者一线治疗采用BTK 抑制剂联合/不联合化疗的患者均获得缓解,这也提示以BTK抑制剂为基础的治疗对携带XPO1基因突变的患者有效。
关于XPO1 基因突变的预后意义,目前尚有争议。Jain等[8]认为XPO1基因突变是低频突变,且对伊布替尼有效,可能并不是CLL 的不良预后因素。而2021年欧洲血液协会(European Hematology Association,EHA)会议报道了一项4 674 例CLL 队列的回顾性研究,则认为XPO1 突变与TTFT 较短相关。2023年Moia等[18]最新研究显示,在仅纳入早期CLL患者的组中,XPO1 突变是TTFT 较短的预测因子。XPO1 突变的CLL 原代细胞的染色质可及性相比XPO1 野生型的CLL 原代细胞更高,可及性增加的染色质区域富含B 细胞受体(B cell receptor,BCR)信号通路中转录因子的结合位点,如NF-κB等。在转录层面上,XPO1 突变的CLL 原代细胞还存在MYB和MIR1HG等基因的过表达,这些基因会刺激BCR 的活性。在本研究中,CLL 患者中位TTFT 为5.2个月,相对较短,这与EHA会议报道和Moia最新研究一致。XPO1基因突变预后意义差异可能与以下4 个因素相关:①XPO1 突变率检测差异;②样本量的差异;③随访时间的差异;④纳入CLL 患者的亚组选择。截至随访结束,6 例R/R XPO1 突变CLL患者中有2例患者在疾病进展后采用BTK抑制剂治疗后仍出现疾病进展,1 例患者发生了Richter 综合症(表2 中P13 患者:IGHV 无突变、β2-MG 升高,分期晚,NGS 可见XPO1、ATM、ARID1A、KMT2D 和FBXW7基因突变),采用BTK 抑制剂联合RTX 并未取得缓解;另1 例患者采用泽布替尼治疗耐药(表2中P12 患者:TP53 异常、β2-MG 升高,分期晚,NGS可见XPO1、TP53、BTK、SF3B1 和MGA 基因突变),后出现中枢浸润。2例患者均在短时间内死亡。因此尽管BTK 抑制剂对多数XPO1 突变患者有效,但仍有部分患者不能克服。此外,本研究注意到,15例CLL 患者中有4 例发生了Richter 综合症,3 例在TN时确诊,其中2例分别采用BTK抑制剂/BCL-2抑制剂联合化疗并序贯自体造血干细胞移植,获得完全缓解。另1 例患者即将启动治疗。1 例在R/R 时确诊,采用BTK抑制剂联合RTX并未取得缓解。
一些研究表明XPO1 是CLL 的有效靶标,且核输出蛋白抑制剂塞利尼索可以延缓CLL 模型小鼠的疾病进展,增加总体生存率[19-20]。也有研究认为,XPO1 突变可能会增加肿瘤细胞对XPO1 抑制剂的敏感性,从而增加XPO1 抑制剂的细胞毒作用[7]。Hing 等[21]研究发现塞利尼索对伊布替尼耐药和携带BTK C481S 位点突变的CLL 模型小鼠有效,同时可以和伊布替尼具有协同作用,可以增加小鼠的总体存活率。对于携带XPO1 突变的R/R 患者,如BTK 抑制剂单药治疗效果不佳,联合XPO1 抑制剂或许是一种新型治疗模式。
目前而言,XPO1 突变在CLL 中似乎并不能提示更差的预后,其为低频突变,且常伴随其他基因突变。在R/R 患者中,XPO1 突变发生率高于TN 患者。对于极其难治患者及发生Richter 综合征的患者,XPO1抑制剂联合靶向治疗或许是治疗的新选择。
猜你喜欢
英语世界(2023年6期)2023-06-30
时代英语·高三(2022年3期)2022-11-10
传染病信息(2022年3期)2022-07-15
大电机技术(2021年3期)2021-07-16
中国生殖健康(2020年2期)2021-01-18
中外文摘(2020年13期)2020-08-01
时代英语·高三(2019年4期)2019-09-03
小学生导刊(2018年13期)2018-06-29
中华老年多器官疾病杂志(2016年9期)2016-04-28
磁共振成像(2015年5期)2015-12-23
