
Shwachman-Diamond 综合征7 例患儿临床特点和基因变异分析
2024-03-16 12:53王瑞芳梁黎黎张开创孙宇宁孙曼青韩连书张惠文顾学范余永国邱文娟
临床儿科杂志 2024年3期
王瑞芳 梁黎黎 张开创 杨 奕 孙宇宁 孙曼青 肖 冰 韩连书 张惠文 顾学范 余永国 邱文娟
上海交通大学医学院附属新华医院 上海市儿科医学研究所 儿内分泌遗传代谢科(上海 200092)
Shwachman‐Diamond 综合征(Shwachman‐Diamond syndrome,SDS,OMIM 260400)是一种罕见的遗传性骨髓衰竭综合征,于1964 年被首次描述[1‐2],其估计发病率为1/76000[3],男女患病率约为1.7∶1[4]。SDS 的临床特征主要为以胰腺外分泌功能不全、骨髓功能障碍和骨骼异常为表现的三联征,此外其还有多种临床表现,包括身材矮小、发育落后、肝脏损害、青春期延迟和牙齿异常等[5]。目前已知SDS的致病基因包括SBDS、EFL1、DNAJC 21或SRP 54,约90%的SDS 由SBDS基因变异所致,该基因编码在核糖体生物合成中起核心作用的蛋白SBDS[6]。另外在核糖体生物发生和蛋白质翻译中发挥作用的EFL 1、DNAJC 21和SRP 54基因的变异也被证实与SDS 发病相关[7]。
SDS是一种累及消化、血液、免疫、骨骼和中枢神经等多系统的疾病,该疾病的临床表型谱很广,在受累个体之间存在显著的表型异质性,为患儿的临床识别和诊断带来挑战。目前国内报道的病例以血液、消化和免疫系统的临床表现多见,但SDS 患儿身材矮小的表现也为其常见的特征,本研究回顾性分析2018—2023年在本院儿内分泌遗传科诊治的7例SDS 患儿的临床资料及其外显子组测序(exome sequencing,ES)结果,旨在探讨中国SDS患儿的临床特点和分子遗传学特征。
1 对象与方法
1.1 研究对象
选择上海交通大学医学院附属新华医院儿内分泌遗传科于2018年1月至2023年9月收治的7例SDS患儿为研究对象。
本研究通过了医院伦理委员会批准(No.XHEC‐D‐2023‐230),所有检查均获得患儿家长或监护人的知情同意。
1.2 方法
1.2.1 SDS诊断标准[3,8]①基因检测:检出SBDS双等位基因致病/可能致病性变异,或其他SDS 相关基因(SBDS、EFL1、DNAJC21或SRP54)的致病/可能致病性变异;②临床表现:血液学特征(至少出现两次):a.中性粒细胞减少(中性粒细胞绝对计数<1.5×109/L);b.其他原因无法解释的贫血或巨红细胞症;c.血小板减少(血小板计数<150×109/L);d.骨髓检查结果异常(骨髓增生异常或白血病或细胞遗传学异常);胰腺特征:a.按年龄校正的胰腺酶类(如血清淀粉酶、血脂肪酶、血清胰蛋白酶原、粪便弹性蛋白酶等)水平降低;b.支持特征:胰腺影像学异常伴脂肪增多症,粪便脂肪排泄增加>72小时;其他支持性证据:a.存在骨骼异常;b.神经认知/行为问题;c.无法解释的身高低于第3百分位数;d.存在被确诊为SDS的一级家庭成员。
1.2.2 临床随访和检查 收集患儿的人口学资料、实验室和辅助检查资料,包括血常规、尿常规、粪便常规、肝肾功能、电解质、凝血功能、血淀粉酶、胰淀粉酶、血脂肪酶、25‐羟维生素D、血胰岛素样生长因子(IGF‐1)、生长激素激发试验;腹部B 超、头颅磁共振成像、双下肢长骨片、骨盆X线片等结果。
1.2.3 基因检测 抽取先证者及其父母静脉血样各3 mL,使用试剂盒(德国 Qiagen 公司)提取基因组 DNA,行家系或者先证者外显子组测序。用外显子组研究检测组套(exome research panel,IDT公司,美国)进行捕获,并用Illumina NovaSeq 6000测序仪(Illumina 公司,美国)对捕获片段进行高通量测序,按照GATK(V3)分析单核苷酸变异和小片段的插入缺失。过滤掉人群数据库中高频的变异(如1000 Genomes Project,Genome Aggregation Database和Exome Variant Server),并且在本地数据库中频率超过1%或超过5%的变异(包括大约6 000 个外显子)也被排除在外,随后通过遗传方式(显、隐性)进一步过滤变异数据。应用外显子组隐马尔科夫模型(eXome Hidden Markov Model,XHMM)在该样本中分析拷贝数变异。经过滤及筛选分析筛选出致病变异位点,按照人类基因组变异协会规则对变异进行命名,参照美国医学遗传学与基因组学学会(American college of medical genetics and genomics,ACMG)的变异解读指南对变异的致病性进行评级[9]。针对检出的阳性变异位点,采用Sanger 测序对先证者及其家庭成员进行验证并判断变异来源。
1.3 统计学分析
采用GraphPad Prism 8.0统计学软件进行数据分析。计量资料符合正态分布的以均数±标准差表示,非正态分布的以中位数M(P25~P75)表示。
2 结果
2.1 一般情况
7例患儿中男3例、女4例,初诊中位年龄为3.0(0.9~4.0)岁,均无家族史。6例患儿均因“身材矮小”首诊于儿内分泌遗传代谢科,余1例因“反复呼吸道感染伴肝功能异常”首诊于儿童感染科,后因存在身材矮小转诊至儿内分泌遗传代谢科,7例患儿的临床特征总结和基因检测结果见表1。
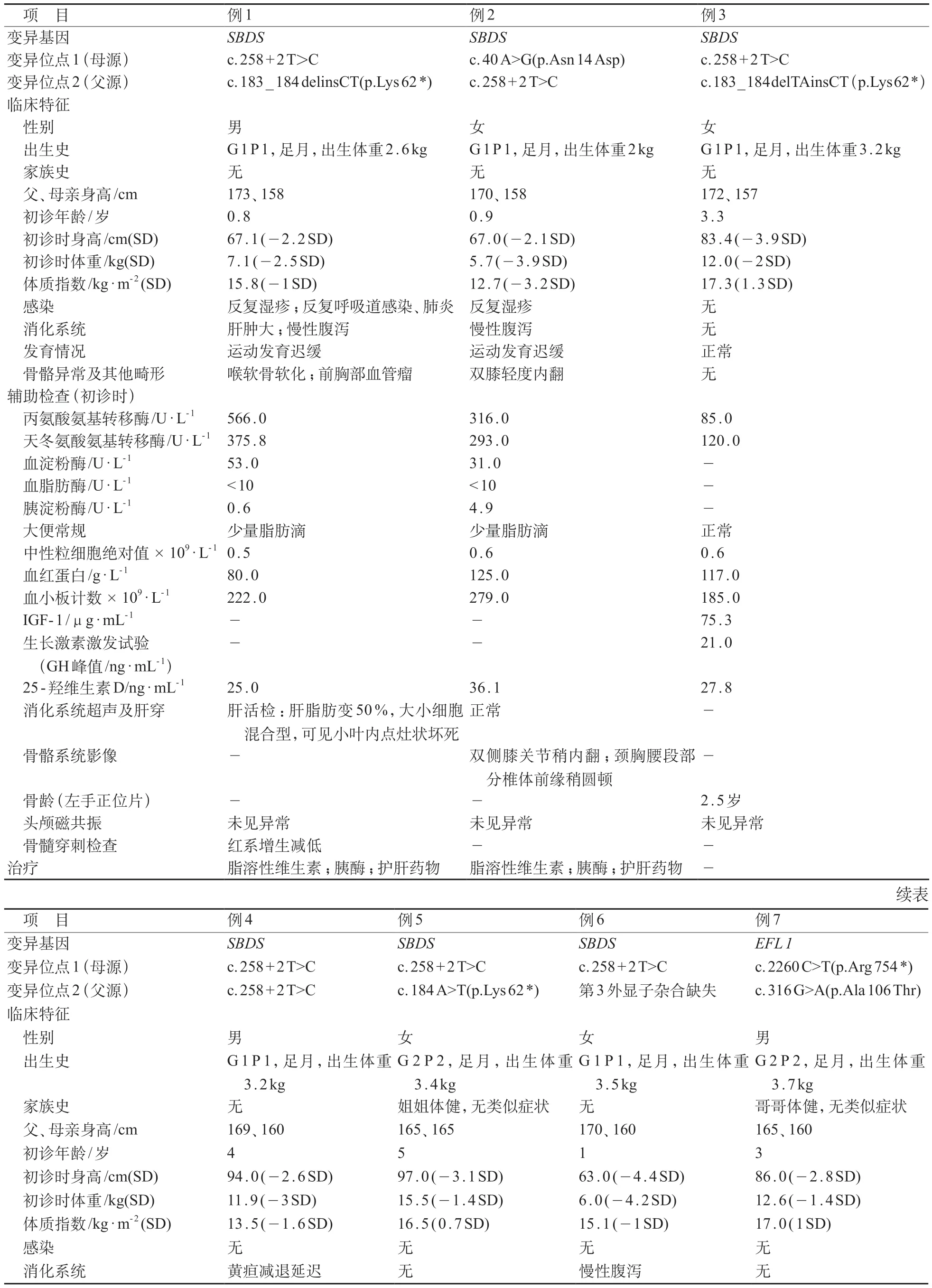
表1 SDS 7例患儿的临床表型和基因型特征总结
2.2 外显子组测序结果
7 例患儿行ES 检测发现6 例患儿携带SBDS双等位基因变异,1 例携带EFL 1双等位基因变异。6例SBDS变异的具体情况见表1(NM_016038.4),共检测出3 种已被报道的突变,包括1 种剪切突变c.258+2 T>C、1 种移码突变c.183_184 delinsCT(p.Lys62*)和1种无义突变c.184A>T(p.Lys62*),另检测到1 种新错义突变c.40 A>G(p.Asn 14 Asp)及第3 外显子的杂合缺失;根据ACMG 遗传变异分类标准与指南,位点c.40 A>G(p.Asn 14 Asp)判定为可能致病性变异。SBDS变异中,c.258+2T>C(7/12,58.3%)和c.183_184delinsCT/c.184A>T(3/12,25%)最常见。
余1例携带EFL1的复合杂合突变[NM_024580.6,c.2260C>T(p.Arg754*)/ c.316G>A(p.Ala106Thr)],其中c.316 G>A(p.Ala 106 Thr)为未报道的新错义突变。根据ACMG 遗传变异分类标准与指南,其为可能致病性变异。
2.3 SBDS缺陷6例患儿的临床表型
6 例携带SBDS变异患儿的临床表型见表1,6例患儿均以矮小(100%)就诊,其中3 例(例1、2、6)伴有慢性腹泻,1 例(例1)伴有反复呼吸道感染。6 例(100%)患儿均出现中性粒细胞减少,其中例1 和例6 伴有贫血。6 例(100%)均有肝酶升高,例1 因肝酶反复增高和肝肿大,行肝脏活检提示肝脂肪变性,可见小叶内点灶状坏死。6 例患儿均存在身材矮小(100%),初诊中位身高83.4 cm(-3.06 SD),例5 完善生长激素激发试验证实为生长激素缺乏症(GHD)。4 例患儿(例1、2、4、5)有骨骼发育异常,包括喉软骨软化、膝内翻、鸡胸、拇指多指畸形等。3 例患儿(例1、2、6)有胰腺外分泌功能不全表现,包括脂肪泻史、胰酶降低或影像学提示胰腺脂肪化等。予加用胰酶制剂、脂溶性维生素等治疗,目前大便次数减少,性状好转。随访肝酶升高的6 例患儿发现,末次随访时例1、2、4 肝酶水平较前下降,例3、5、6 随访肝酶基本正常。例5 确诊GHD 后开始使用生长激素治疗,随访数据缺失。
2.4 EFL1缺陷1例患儿的临床表型
1例携带EFL1变异的患儿临床表型见表1。例7以身材矮小(初诊身高86cm,-2.84SD),走路姿势异常就诊,体检发现双膝外翻、左腿明显弯曲及扁平足,影像检查提示存在脊柱、骨盆、双下肢多发骨质异常,多发性骨骺(股骨/膝关节/踝关节/髋臼)发育不良。患儿无复发性感染、胰腺外分泌功能不全表现和脂肪泻的症状,无肝功能异常及血液系统受累表现。患儿存在矮小及血IGF‐1偏低,且经生长激素激发试验证实为GHD,并接受生长激素注射治疗,末次随访时生长激素治疗时长为15个月,身高提高8.6 cm(0.24SD)。
3 讨论
SDS是一种可累及全身多系统多器官的遗传性综合征,除外基因诊断,根据2011 年国外SDS 的共识,临床诊断主要基于中性粒细胞减少和胰腺外分泌功能不全的特征[8]。但既往报道的国外不同人群SDS临床表现具有广泛的多样性[11‐16],部分患儿以矮小、肝功能异常等其他症状为首发表现,而缺乏经典的胰腺外分泌功能不全和骨髓功能障碍的表现,导致临床容易漏诊。国内目前报道40 余例SDS 患者[17],多为个例报告,临床表型主要累及消化、免疫和血液等系统,其中携带SBDS基因变异的41 例SDS 患儿的中性粒细胞减少症、胰腺外分泌功能不全和骨骼异常比例分别为92.7%、65.8%和41.5%。本研究的7例患儿临床表型差异也较大,7例患儿均存在生长迟缓,其中性粒细胞减少症、外分泌胰腺功能不全和骨骼异常的比例分别为85.7%、42.9%和85.7%。与国内外的相关人群报道比较,中性粒细胞缺乏(59.9%~100%)和胰腺外分泌功能不全(63.6%~100%)在大多数人群中报道的比例相仿,均较高,但骨骼异常在各人群中的比例差异较大(8.8%~80.6%)[11‐16],并常伴有其他多个系统的合并症状。因此,加深对SDS 综合征多样性表型的认识非常关键,疾病早期缺少经典症状的患者也不能排除SDS的诊断,需完善基因检测进一步明确诊断。
SDS 是一种核糖体生物发生障碍性疾病,国外报道约90%的患者携带位于染色体7q11上的SBDS基因的双等位基因变异,该基因编码的同源SBDS蛋白被认为在核糖体生物合成和RNA 代谢中发挥重要作用[7]。约10%的SDS 患者未携带SBDS基因的致病变异,近年研究提示另外3 个与核糖体组装或蛋白质翻译相关的基因(EFL 1、DNAJC 21和SRP 54)也为SDS 综合征的候选基因[18‐20]。国外共报道了63例SDS患者携带此3个基因的变异,其中SRP54变异33例(52%)、DNAJC21变异 17例(27%)和EFL 1变异13 例(21%)[21]。而目前国内共报道了42 例明确基因变异的SDS 患者,其中41 例携带SBDS变异(97.6%),1例携带SRP54变异(2.4%),尚无DNAJC21和EFL1变异的报道[17]。本研究共7例基因确诊的SDS患儿也以SBDS变异为主,分别为SBDS变异6例(85.7%)和EFL1变异1例(14.3%)。
导致SDS 致病最常见的SBDS变异具有狭窄的基因型谱,占致病等位基因76%以上的3 种最常见突变位点为c.258+2 T>C、c.183_184 delinsCT 和(c.183_184delinsCT;258+2T>C)[11]。本研究7例SDS 患儿中6例携带SBDS基因的变异,其中5例携带复合杂合突变,1例携带纯合突变(c.258+2T>C),以热点突变c.258+2 T>C 和c.183_184 delinsCT/c.184A>T为主,分别占58.3%和25%。其中,例1、3、5 患儿均携带复合杂合突变,其中1 条等位基因上携带c.258+2 T>C 突变,另1 条等位基因上携带编码相同氨基酸改变的突变c.183_184delinsCT或c.184A>T,但临床表型有较大差异。例1表现有典型的中性粒细胞减少伴反复湿疹和呼吸道感染、胰腺外分泌功能障碍伴脂肪泻、肝酶明显升高和肝肿大、生长及发育落后,及喉软骨软化等骨骼畸形;例3和例5则以生长迟缓为主,伴中性粒细胞减少和轻度肝酶升高,无胰腺外分泌功能障碍,例5 伴发左拇指多指畸形,此2 例患儿缺乏经典的SDS 临床症状,经基因检测才确定该疾病的诊断。例2 携带剪接突变c.258+2 T>C 与1 个新错义突变c.40 A>G(p.Asn14Asp),临床表型为中性粒细胞减少伴反复湿疹,胰腺外分泌功能障碍伴脂肪泻,肝酶升高,生长及发育落后,及以双膝轻度内翻为表现的骨骼异常;而例4 携带c.258+2 T>C 纯合突变,表现为典型的中性粒细胞减少、黄疸减退延迟和肝酶升高、生长及发育落后,骨骼畸形有牙齿稀和鸡胸等。可见SBDS基因不同变异可引起不同的表型,相同的变异位点其并发的临床表型也呈现异质性,基因型和临床表型无明确的相关性,这更凸显了对临床表型全面评估和准确基因检测的重要性。
在余3 个SDS 相关的非SBDS基因中,EFL 1变异导致的SDS目前国外共报道了13例患儿[20,22‐24],国内尚无相关报道。该基因定位于15q25.2,编码三磷酸鸟苷酶(GTP酶)类延伸因子1(EFL1),为核糖体转位酶延长因子2(EF‐2)的同源物[6,21]。研究显示在核糖体60S亚基成熟的最后一步,SBDS与GTP酶EFL1合作催化核糖体抗结合因子(eIF 6)的释放并激活翻译,EFL1变异阻止了晚期细胞质60S亚基中eIF6的释放,进而损害核糖体亚基的连接并减弱整体的翻译,这为其可能的致病机制[22]。本研究例7携带EFL1的2种变异,其中无义突变(c.2260C>T/p.Arg 754*)位于结构域Ⅳ,其对于EFL 1 在体内的功能是必需的[25]。而另1 个错义突变(c.316 G>A/p.Ala 106 Thr)既往无报道,位于第Ⅰ结构域,该结构域含有参与鸟嘌呤核苷酸(GTP)结合和催化的特征性G 1~G 5 结构基序,这个变异毗邻G 3 基序,该结构域在GTP 酶从非活性状态(GDP)交替到活性状态(GTP)的功能循环期间经历大的构象变化,对GTP酶活性作用的发挥非常重要[26]。根据ACMG标准预测EFL 1的这个变异为致病性变异,且在多种物种间保持高度保守的氨基酸,故该变异可判定为损害EFL 1 功能的变异,且经家系连锁分析也符合遗传模式,该患儿的EFL 1基因型和其SDS 的临床表型符合。国外报道的13例EFL1变异的SDS患者中,中性粒细胞减少症、胰腺外分泌功能不全和骨骼发育异常的比例分别为100%、77%和92%[20,22‐24],而本研究报道的EFL 1变异主要表现为矮小和骨骼发育异常,无胰腺外分泌功能障碍及血液系统受累,与国外报道有较大不同。这提示EFL 1变异引起的SDS 表型也存在异质性,对于存在生长落后及骨骼发育异常的患儿,需警惕SDS的可能。
本研究7例SDS患儿中6例均因“身材矮小”首诊于儿内分泌遗传代谢科,余1例因“反复呼吸道感染伴肝功能异常”首诊于儿童感染科,后因存在身材矮小转诊至儿内分泌遗传代谢科。经评估内分泌系统相关表型除身材矮小外,4 例患儿(例4~7)有血IGF‐1水平偏低,其中2例(例5和例7)同时伴生长激素缺乏,未发现其他内分泌相关异常。血IGF‐1水平的降低可能与生长激素分泌不足、营养不良、慢性肝病等慢性消耗性疾病等因素相关[27]。例5和例7无慢性腹泻表现,肝功能为轻度异常到正常范围,故其血清IGF‐1水平的降低考虑主要由生长激素分泌不足引起,余2例患儿血IGF‐1水平偏低则可能受慢性腹泻和营养不良等因素影响,可在后续随访继续监测。既往相关研究评估了来自波兰的19例SDS患儿内分泌功能情况,13 例(63%)SDS 患儿表现出一种或多种内分泌疾病,其中7 例(37%)为生长激素缺乏(GHD),并对该7 例患儿进行了2 年的生长激素(rGH)治疗,证实rGH治疗可显著改善SDS患儿的身高标准差和生长速率,且未观察到明显的不良反应[28‐29]。因SDS 具有较高的向髓样肿瘤(骨髓增生异常综合征/急性髓系白血病)转化的风险(10%~30%)[21],故权衡rGH 治疗的个体化获益与风险非常重要。目前对SDS合并GHD患儿的生长激素治疗数据较少,本研究的2例SDS合并GHD患儿也进行了生长激素治疗,但目前治疗时间均较短,需要进一步研究来分析生长激素治疗SDS患儿的长期效果和安全性。
综上所述,SDS 是一种罕见的引起多系统受累的遗传综合征,其有经典的三联症表型,但临床症状变异性较大,部分患儿因临床表型不典型易造成漏诊或延迟诊治。本研究总结了7 例SDS 患儿的基因型和临床表型特点,其中1例EFL1变异患儿为在中国人群中首次报道,丰富了SDS 的疾病谱。对出现生长迟缓合并中性粒细胞减少、胰腺外分泌功能障碍、骨骼畸形等多种症状的患儿,应怀疑 SDS,完善基因检测以免漏诊。对基因确诊的患儿,应行多系统随访和多学科综合治疗,其中内分泌也为SDS常累及的系统之一,应全面评估内分泌功能情况,以期尽早干预,改善预后。
猜你喜欢
中老年保健(2021年5期)2021-12-02
中老年保健(2021年5期)2021-08-24
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年6期)2020-02-01
中国生殖健康(2018年6期)2018-11-06
科学与财富(2018年8期)2018-05-09
小布老虎(2017年1期)2017-07-18
中华胰腺病杂志(2015年5期)2015-12-08
中国医疗美容(2015年4期)2015-04-27
实用器官移植电子杂志(2015年5期)2015-04-03
