
The role of innate immunity in diabetic nephropathy and their therapeutic consequences
2024-03-21 05:51MinYangChunZhang
Min Yang, Chun Zhang
Department of Nephrology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, China
Keywords:Innate immunity Diabetic nephropathy Inflammation Toll-like receptor Inflammasomes
A B S T R A C T Diabetic nephropathy(DN)is an enduring condition that leads to inflammation and affects a substantial number of individuals with diabetes worldwide.A gradual reduction in glomerular filtration and emergence of proteins in the urine are typical aspects of DN, ultimately resulting in renal failure.Mounting evidence suggests that immunological and inflammatory factors are crucial for the development of DN.Therefore, the activation of innate immunity by resident renal and immune cells is critical for initiating and perpetuating inflammation.Toll-like receptors (TLRs) are an important group of receptors that identify patterns and activate immune responses and inflammation.Meanwhile, inflammatory responses in the liver,pancreatic islets,and kidneys involve inflammasomes and chemokines that generate pro-inflammatory cytokines.Moreover, the activation of the complement cascade can be triggered by glycated proteins.This review highlights recent findings elucidating how the innate immune system contributes to tissue fibrosis and organ dysfunction, ultimately leading to renal failure.This review also discusses innovative approaches that can be utilized to modulate the innate immune responses in DN for therapeutic purposes.
1.Introduction
Diabetic nephropathy (DN) is a prevalent and severe diabetesrelated complication that frequently results in terminal renal dysfunction and increases mortality[1,2].DN is the leading cause of end-stage renal disease(ESRD)and affects approximately 20%-50%of individuals with diabetes.This condition not only imposes a financial burden but also significantly reduces the overall standard of living of affected individuals [3].The clinical manifestations of DN typically include a decrease in the glomerular filtration rate and elevated urinary protein levels.The structural pathology of DN involves thickened basement membranes, expanded glomerular mesangial cells,podocyte loss,glomerulosclerosis,and destruction of endothelial cells.Patients with DN often present with tubular hypertrophy in the early stages, which can progress to glomerular and tubulointerstitial fibrosis.Several factors are believed to be involved in the initiation and progression of DN.These include inflammation,metabolic disorders,and hemodynamic changes[4].Immunological damage is a prevalent subject in DN-related research.In 1991, Hasegawa et al.[5] first proposed that inflammatory factors may play a role in DN.Interestingly,evidence linking inflammation to DN progression is increasing[6,7].Inflammation is a prominent factor in the advancement of DN and is characterized by elevated infiltration of inflammatory cells, release of proinflammatory cytokines, production of chemokines, and tissue damage.In individuals with diabetes, elevated glucose levels can disrupt metabolism and circulation, leading to the activation of innate immune response in various types of kidney cells,including podocytes, tubular epithelial cells (TECs), and mesangial cells.Several molecules are upregulated and involved in the progression of DN.These include inflammatory cytokines such as interleukin(IL)-1, IL-8, and tumor necrosis factor-α (TNF-α) [8,9] as well as chemokine(C-C motif)ligand 2(CCL-2)[10],intercellular adhesion molecule 1 (ICAM-1) [11], transforming growth factor-β (TGF-β),and vascular endothelial growth factor[12].These findings suggest possible therapeutic targets for the treatment of DN.Moreover,the accumulation and stimulation of macrophages in the kidneys are notably correlated with albuminuria and renal fibrosis in individuals with diabetes[13].
Innate immunity plays a crucial role not only in fighting bacterial and viral infections but also in activating adaptive immunity.The innate immune system rapidly recognizes and eliminates injury caused by pathogen invasion.Pathogens activate pattern recognition receptors (PRRs) to trigger an initial innate immune response by detecting specific pathogen-associated molecular patterns (PAMPs).This is the initial step in recognition of and response to microbial invasion.Furthermore, the innate immune system detects internal damage signals by recognizing damageassociated molecular patterns (DAMPs) [14].DAMPs are generated by cells that have been damaged or are in the process of dying.These molecules include ATP and DNA as well as uric acid crystals.Asbestos also contributes to the release of DAMPs.Additionally,ultraviolet radiation contributes to the generation of DAMPs [15].Strong evidence supports the contribution of innate immunity to the inflammatory pathways associated with diabetes [16].Germline-encoded receptors called PRRs mediate innate immune responses [17].This review focuses on the latest progress in understanding the functional significance of toll-like receptors(TLRs),nucleotide-binding oligomerization domain (NOD)-like receptors(NLRs), chemokines, and the complement system in the development of DN.
2.TLRs signaling
TLRs, the main constituents of the innate immune system,trigger inflammatory pathways.TLRs can identify both PAMPs and DAMPs in the extracellular environment and endosomes.Currently,the human TLR family comprises ten members.It is worth noting that TLRs are not only found in immune cells,such as macrophages,dendritic cells, lymphocytes, and neutrophils, but also in various non-immune cells, including endothelial cells, TECs, mesangial cells, fibroblasts, and podocytes [18].Following their synthesis within the endoplasmic reticulum, TLRs can be transmitted to the cellular surface or endosomes.TLRs that are present on the cell surface can recognize PAMPs,such as lipopolysaccharides(LPS)and bacterial flagella, as well as DAMPs released from damaged or stressed cells,such as high-mobility group protein B1(HMGB1)and heat shock proteins (HSPs).TLR1, TLR2, TLR4, TLR5, and TLR6 are specifically responsible for this process [19].Endocellular TLRs,including TLR3,TLR7,TLR8,TLR9,and TLR10,identify nucleic acids derived from microorganisms [20].The downstream signaling pathways activated by TLRs can be categorized into two types:those dependent on myeloid differentiation factor 88(MyD88)and those independent of MyD88 [21].Many TLRs trigger MyD88-dependent pathways by binding to their ligands, which results in the synthesis of inflammatory cytokines.However, alternative signaling routes that do not involve MyD88 (primarily TLR4 and TLR3) are responsible for the initiation of interferon (IFN)-β production [22].Activation of TLRs results in the recruitment of adaptor proteins such as MyD88, TIR domain-containing adaptor inducing interferon(TRIF),TIR domain-containing adaptor protein,and TRIF-related adaptor molecule[23].Adaptor molecules initiate downstream signaling pathways that activate nuclear factor-κB(NF-κB) and transcription factors (Fig.1).This ultimately leads to the synthesis of chemokines and pro-inflammatory cytokines[24,25].
Pathogens such as bacteria can invade the host during infection by breaching the epithelial barrier and triggering the activation of macrophages through the recognition of PAMPs by TLRs.The activation of TLR leads to the generation of pro-inflammatory cytokines, mobilization of neutrophils, and synthesis of antimicrobial agents, including reactive oxygen species (ROS), peptides, and leukotrienes.Macrophages and neutrophils engulf pathogens to reduce infection[26].When damaged or dead cells release DAMPs,the immune system is triggered to differentiate between dangerous and non-threatening conditions.Thus, the activation of TLRs from DAMPs may give rise to sterile inflammation.However, the physiological processes involved in cell death are typically cleared.Increasing evidence suggests that TLRs may be involved in numerous non-infectious inflammatory ailments such as diabetes,atherosclerosis, and chronic kidney disease [27-30].
2.1.TLR signaling in DN
Numerous clinical and experimental studies have demonstrated an association between TLRs and the pathophysiological processes of DN [31].TLR signaling proceeds via two types of pathways:MyD88-mediated and MyD88-independent pathways.The former leads to the activation of NF-κB and multiple mitogen-activated protein kinases (MAPKs), which in turn trigger the activation of multiple genes that contribute to inflammatory responses.The latter triggers IFN production and induces a cellular antiviral response.TLR3 specifically triggers the TRIF-mediated signaling pathway, which ultimately leads to the activation of interferon regulatory factor (IRF)-3.IRF-3 induces the expression of type I IFNs.Additionally,this pathway results in the indirect upregulation of IFN-associated genes, such as IFN-inducible protein 10 and inducible nitric oxide synthase.Furthermore, TLR3 triggers the activation of NF-κB, inducing the synthesis of inflammatory cytokines including IL-1β,IL-6,and TNF.TLR4 activates both pathways,triggering NF-κB activation and inducing type I IFN production.In contrast, all other TLRs exclusively activate the MyD88-dependent pathway.Activation of TLR signaling is associated with increased production of pro-inflammatory mediators,which contribute to the development of sustained chronic inflammation in diabetes.Studies have demonstrated a noteworthy increase in the protein levels of TLR2 and TLR4, which corresponded to the enhancement of their respective ligands and MyD88-dependent expression of NF-κB in monocytes extracted from patients with type 1 diabetes(T1DM)and type 2 diabetes(T2DM).Most studies of chronic renal injury and TLRs have primarily examined TLR2 and TLR4 expression.Nevertheless,there is evidence that the levels of downstream molecules of the TLR3 pathway, such as pro-inflammatory cytokines, are elevated in patients with DN.Among all TLRs, TLR2 and TLR4 have been extensively studied in the development of DN.TLR2 and TLR4 play crucial roles in various kidney disorders such as glomerulonephritis, renal ischemia, kidney transplantation, and diabetic tubular inflammation [32-36].According to recent evidence, TLR2 and TLR4 significantly contribute to persistent inflammation in DN[37].Both TLR2 and TLR4 are highly expressed in circulating monocytes of individuals with diabetes [27].Additionally,downstream targets,such as MyD88,TRIF,phosphorylated IL-1 receptor-associated kinase, and NF-κB, are notably upregulated.It is also believed that NF-κB may play a role in other processes, such as the accumulation of advanced glycation end products (AGEs) and activation of the renin-angiotensin system(RAS) pathways, protein kinase C, and oxidative stress in DN[38-41].When endothelial cells are exposed to various concentrations of glucose, the levels of TLR2 and TLR4 increase, followed by the increasing levels of NF-κB,IL-8,CCL-2,ICAM-1,and vascular cell adhesion molecule-1[42].In animal models of T1DM,including non-obese diabetic mice and streptozotocin(STZ)-induced diabetic mice,there appears to be an increase in the expression of TLR2 and TLR4 as well as the activation of downstream signaling pathways[43-45].Based on clinical evidence,TLR2 and TLR4 ligands such as endotoxins, HSP60, and HMGB1 are more abundant in patients with T1DM (Table 1) [37,46].MyD88 is a crucial intracellular adaptor protein that plays a bridging role between TLRs activation and the subsequent inflammatory response.Mice lacking TLR2,TLR4, or MyD88 show less unilateral ureteral obstruction associated with interstitial fibrosis [47].Mice lacking MyD88 and fed a high-fat diet exhibit elevated insulin,leptin,and cholesterol levels,which are associated with insulin resistance and the progression of severe T2DM [48].After low-dose STZ treatment, the mice exhibited reduced islets and impaired glucose tolerance [49].

Fig.1.Signaling pathways of Toll-like receptors(TLRs).Endogenous ligands triggered by diabetic state can be recognized on the cell surface through TLR2 and TLR4 upon exposure to diabetic stimuli.MyD88:myeloid differentiation factor 88;TIRAP:TIR domain-containing adaptor protein;TRIF:TIR domain-containing adaptor inducing interferon;TRAM:TRIFrelated adaptor molecule;TRAF:TNF receptor-associated factor;TAK:TGF-β activated kinase;TAB:TAK1-binding protein;IKK:inhibitor of kappa B kinase; NEMO:NF-κB essential modulator;TBK:TANK-binding kinase 1;IRAK:interleukin-1 receptor-associated kinase;MAPK:mitogen-activated protein kinase;MD2:myeloid differentiation 2;NF-κB:nuclear factor-κB; IRF: interferon regulatory factor; IFN: interferon; AP-1: activator protein-1.
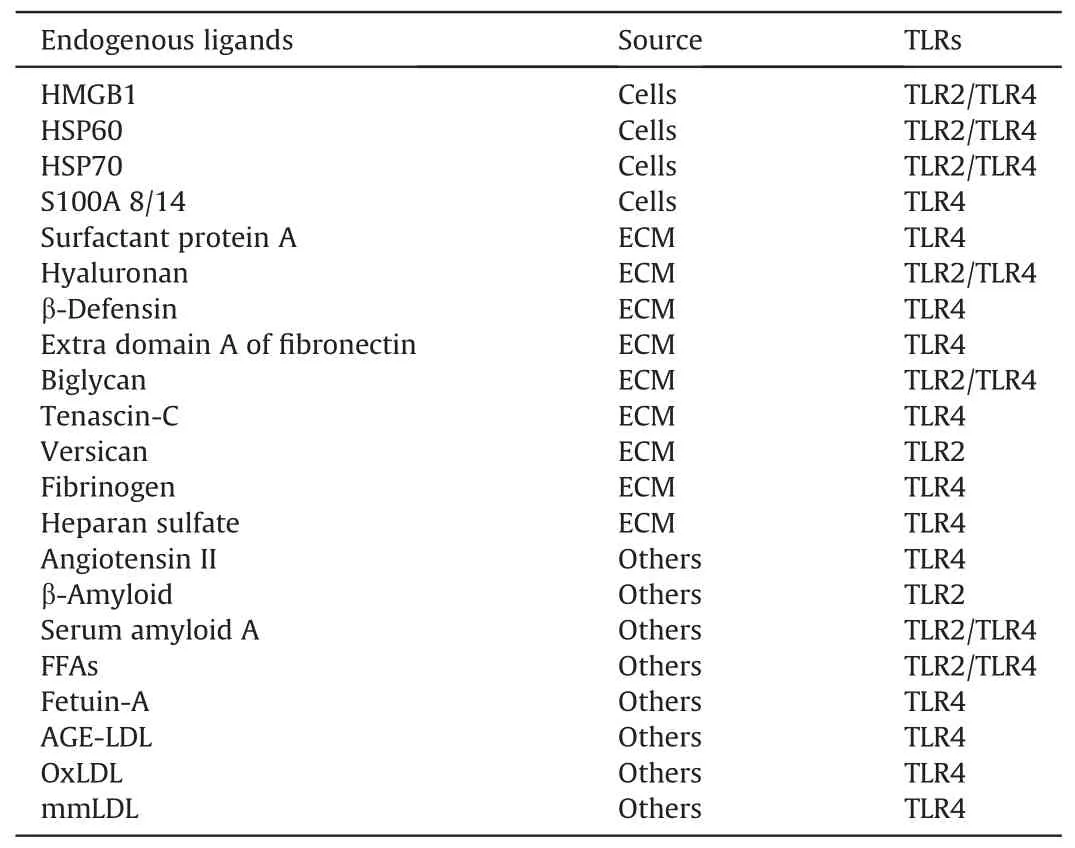
Table 1 Proposed endogenous ligands for Toll-like receptors(TLR)2/4 in diabetic-associated disorders [37,46].
2.1.1.TLR2 in DN
High TLR2 expression has been reported in the glomeruli and renal tubules of diabetic mice.TLR2,MyD88,and pro-inflammatory cytokines are more readily expressed in the leukocytes of patients with T1DM,suggesting that TLR2 contributes to inflammation[50].In cultured NRK-52E cells, TLR2 mRNA expression was strongly influenced by glucose concentration [51-53].Using the STZinduced diabetes model, the absence of TLR2 led to a significant decrease in inflammation associated with diabetes.The levels of pro-inflammatory cytokines and chemokines decreased noticeably due to a substantial decline in the activity of NF-κB, MyD88, and phosphorylation of interleukin-1 receptor-associated kinase 1[44].In addition, unlike wild-type diabetic mice, TLR2-/-STZ mice exhibited noticeable improvement in albuminuria, an increase in podocyte number, and decreased M1 macrophages in the kidney.Moreover, TLR2-/-STZ mice experienced an increase in nephrin and podocin and a decrease in TGF-β and laminin levels [54].Similar results were obtained when TLR2 was knocked down in cells using short interfering RNA (siRNA) under hyperglycemic conditions[55].Therefore,although TLR4 expression is increased in the diabetic milieu, TLR2 deficiency has a profound effect on MyD88-dependent signaling.
2.1.2.TLR4 in DN
One of the earliest recognized and most intensively studied TLRs in humans is TLR4.Renal mesangial cells, tubular epithelial cells,and podocytes exhibit significant expression of TLR4.TLR4 plays a significant role in inducing inflammation and fibrosis in the kidneys[56].Previous studies have demonstrated that TLR4 is actively involved in renal ischemia-reperfusion injury [34],glomerulonephritis [57], and tubulointerstitial nephritis [58], as well as in the occurrence and development of DN [36].Under proteinuric conditions, plasma macromolecules such as fibrin or fibrinogen that appear in the Bowman's space are capable of inducing podocyte cytokines via TLRs, which accelerate podocyte damage [59].TLR4 is upregulated in inflammatory glomerular diseases,where it is constitutively expressed by podocytes and may be used to mediate glomerular injury by chemokines [60].In addition, LPS contributes to the progression of proteinuria by altering the actin cytoskeleton in podocytes via the TLR4 signaling pathway [61].These findings suggest that TLR4 activation in podocytes is critical for DN.
In diabetic biopsy samples from patients with T2DM, excessive expression of tubular TLR4 showed a positive correlation with interstitial macrophage infiltration and HbA1clevels,and a negative correlation with renal function [36].The study confirmed heightened TLR4 protein and mRNA expression in both glomeruli and tubules in renal biopsy samples obtained from patients diagnosed with T2DM [62].
In diabetic mice subjected to experiments, increased TLR4 activity and amplified expression of the NF-κB p65 subunit in the kidney cortex were observed[63].When exposed to high levels of glucose and non-esterified fatty acids, podocytes and adipocytes exhibit increased expression of TLR4 and synthesis of downstream proinflammatory cytokines.This finding implies that the activity of TLR4 is vital for mediating inflammation in DN [64,65].Under elevated glucose conditions, the initiation of ROS production and NF-κB signaling activation was linked to the upregulation of TLR4 expression in cultured podocytes [66].In addition, hyperglycemia stimulated TLR4 expression in glomerular renal endothelial cells of T1DM and T2DM mice[67,68].TLR4 but not TLR2 was increased in mesangial cells as a result of high glucose,supporting the dominant role of TLR4 in diabetic renal inflammation [67].DN-associated renal inflammation, podocytopathy, and fibrosis could be improved by depleting TLR4 [69].Moreover, TLR4-/-mice treated with STZ exhibited improved renal function and reduced albuminuria.This advantageous characteristic was accompanied by a considerable reduction in the expression of CCL-2 and infiltration of macrophages into the tubulointerstitium [70].
Several studies have shown that TLR4 activates inflammation and renal fibrosis in DN patients [37].The initiation of a TLRmediated immune response is critically dependent on endogenous ligands.NF-κB activation in immune and renal cells is mediated by TLR4 activation through MyD88-dependent and independent pathways [71].Dendritic cells, macrophages, and necrotic cells release an endogenous ligand of TLR4, HMGB1, into the extracellular fluid during inflammation [72].High levels of HMGB1 and HSPs have been observed in the serum and renal tissues of individuals with diabetes [73].Vitro experiments have shown that mesangial and proximal tubular cells exhibit increased HMGB1 expression under high glucose conditions [74,75].However, glycyrrhizin, an HMGB1 inhibitor, was effective in reducing NF-κB activation,oxidative stress,and inflammation caused by high glucose levels in renal cells [76,77].Additionally, serum of db/db mice could induce the expression of HMGB1 as well as epithelial to mesenchymal transition and apoptosis in cultured podocytes,while inhibiting HMGB1 via siRNA may hinder these effects caused by serum [78].Podocytes of db/db mice were similarly protected by HMGB1 short hairpin RNA (shRNA) therapy.
HSP70 has also been implicated in the pathogenesis of DNassociated inflammation.The expression of HSP70 was upregulated in proximal tubular cells upon exposure to albumin.The TLR4 pathway-mediated synthesis of pro-inflammatory cytokines was diminished in STZ-induced diabetic mice by suppressing HSP70 activity with a transcriptional inhibitor, such as KNK437, or an antibody that neutralizes HSP70 [79].TLR4 knockout had a protective effect on the kidneys, whereas TLR2 knockout had no significant effect.Proteinuria induces inflammation and kidney damage by activating the HSP70-TLR4 pathway.Thus, the expression and signal transduction of TLR4 are enhanced in response to high glucose levels, causing DN to progress more rapidly [80].
2.1.3.TLR4 and RAS in DN
Few studies have addressed the possibility of activating the TLR pathway and dysregulating the RAS components that cause renal inflammation[81].Historically,RAS has been widely acknowledged as a vital system for humoral regulation in the human body.It comprises several peptide hormones and related enzymes[82].The following molecules are involved in the conventional RAS: angiotensin I (Ang I), angiotensin II (Ang II), angiotensin I-converting enzyme (ACE), angiotensin I-converting enzyme-2 (ACE2), angiotensinogen, renin, aldosterone, Ang II type 1 receptor (AT1R), and Ang II type 2 receptor (AT2R).In addition to mediating vasoconstriction and blood pressure, the RAS contributes to cell proliferation, inflammation,oxidative stress,and renal fibrosis [83,84].The pathogenic process may involve the indirect modulation of TLR4 along with its direct pro-inflammatory properties through RAS components.Many studies have demonstrated that Ang II enhances TLR4 expression and stimulates TLR4-mediated signaling pathways in multiple organs and cell populations, contributing to proinflammatory effects.Ang II enhances TLR4 expression in mouse mesangial cells and podocytes, which contributes to renal inflammation [85].Ang II and TLR4 exhibited a synergistic effect in inducing inflammation in mesangial cells of rats exposed to high glucose conditions.These findings suggest that RAS activation may be an additional mechanism contributing to the activation of TLR4 in DN[86].Research has shown that Ang II plays a significant role in the regulation of the TLR4 pathway in DN.However,high glucoseinduced TLR4 overexpression was later shown to affect Ang II expression.Local activation of RAS and production of Ang II are mediated by TLR4 via MAPK-dependent pathways.Blocking TLR4 significantly reduces renal RAS activity in DN [87].When high glucose stimulation and hypertension are present,RAS components and TLR4 signaling pathways may interact to regulate the onset and progression of DN.
2.2.Targeting TLR signaling in DN
TLR2/4 signaling is a promising therapeutic target for DN as it can trigger and sustain chronic inflammation, albuminuria, and fibrosis.Increasing experimental data suggests that renal protection may result from the suppression of TLR signaling,particularly that of TLR2 and TLR4.The monoclonal antibody, OPN-305, which targets TLR2, is currently undergoing phase II clinical trials to prevent delayed graft function [88].The TLR2 functional aptamer AP177 was identified as being capable of significantly suppressing NF-κB and the production of cytokines[89].In contrast,treatment with CRX-526 markedly inhibited albuminuria, parenchymal inflammation,and collagen deposition,indicating the effectiveness of CRX-526 therapy for inhibiting TLR4 signaling in eNOS-deficient mice with DN [70].During the phase III trial, the development of TAK242, a TLR4 antagonist, was terminated [90].However, it was shown that TAK242 suppressed the HMGB1/TLR4 signaling,resulting in decreased binding of NF-κB in human tubular epithelial cells [74].The drug GIT27, which inhibits TLR2/4, is currently in Phase I clinical trials.GIT27 exerts a renoprotective effect by modulating TLR4 signaling [63].Furthermore, miRNAs can be utilized in additional strategies for targeting adapter proteins that play a role in TLR signaling[88,91,92].An additional promising strategy is to modify the interaction between TLRs and their intrinsic ligands,including HMGB1[93].Although current data indicate that TLRs, particularly TLR4, potentially regulate inflammation in DN,additional studies are necessary to confirm these findings.Blocking the classical receptor for endotoxins, TLR4, may pose security concerns in certain infectious and immunological scenarios,because it may have adverse effects.A deeper understanding of the pathophysiology of TLR signaling could assist in the development of targeted therapies for inflammation and fibrosis in DN.
3.Involvement of NLRs
NLRs are a type of PRR primarily located in the cytoplasm.Both intracellular PAMPs and DAMPs were identified.At present,22 and 35 NLR genes have been identified in humans and mice, respectively [94,95].All NLRs contain a central nucleotide binding and oligomerization (NACHT) domain.In addition, most NLRs contain ligand-sensing C-terminal leucine-rich repeat domains [96,97].NLRs feature an effector domain at their N-terminus that facilitates downstream protein interactions.This may be an acidic transactivation domain, caspase recruitment domain (CARD), pyrin domain (PYD), or baculoviral inhibitory repeat (BIR)-like domain[98].Four distinct subfamilies of NLRs exist based on their N-terminal domains: NLRAs, NLRCs, NAIPs (also known as NLRBs), and NLRPs.The solitary member of the NLRA subfamily is class II transactivator (CIITA).The NLRB subfamily proteins contain a BIRlike domain at the N-terminus.The NLRC subfamily, comprising NOD1, NOD2, and NLRC3-5, is distinguished by the presence of a CARD domain.Finally,the NLRP subfamily,including NLRP1-14,all feature a PYD domain [99].
NOD1 is present in renal TECs and many other cells and organs in both humans and mice.In humans,glomerular endothelial cells and TECs can detect NOD2, whereas in mice, mesangial cells,podocytes, and TECs can detect their expression [100].Both NOD1 and NOD2 can bind to receptor-interacting protein 2 kinase via CARD-CARD interactions.This ultimately activates the NF-κB or MAPK pathway, leading to the production of inflammatory cytokines such as IL-6, IL-1β, and TNF-α [97,99].NOD1 and NOD2 interact with ATG16L to facilitate autophagy [101].In addition,NLRP1,NLRP3,NLRP6,NLRP12,and NLRC4 can assemble and create inflammasome complexes that stimulate caspase 1, ultimately resulting in the release of IL-1β and IL-18 [102].It is becoming increasingly evident that NLRs,including NOD1,NOD2,and NLRP3,contribute significantly to DN development.
3.1.NOD1 and NOD2
The development of DN may be aided by NOD1 receptors through activation of the NOD1-RICK-NF-κB pathway, which is implicated in the progression of insulin resistance [103].NOD1 deficiency protects against insulin resistance induced by a high-fat diet, which is consistent with the effects of NOD1 on obesityinduced insulin resistance and inflammation [104].Additionally,it has been proven that NOD2 is present in TECs, podocytes,mesangial cells, endothelial cells, and inflammatory cells [100].In both mice and humans with DN, there is a notable elevation in NOD2 expression within the kidneys, which is associated with DN severity.In vitro, studies suggest that NOD2 receptors induce inflammation and insulin resistance [105].Glomerular endothelial cells play a crucial role in limiting proteinuria and subsequent renal dysfunction.Endothelial cells in the glomerulus play a critical role in preventing proteinuria and renal impairment [106], and NOD2 may boost the endothelial-to-mesenchymal transition(EndMT)via the mitogen-activated protein(MEK)/extracellular signal-regulated kinase (ERK) pathway.Suppression of NOD2 expression led to a significant decrease in high glucose-induced EndMT [107].Moreover,NOD2 may be induced by high glucose,AGEs,TNF-α,and TGF-β1 levels in podocytes[105].In mice with DN,NOD2 deficiency results in decreased suppression of nephrin expression, leading to attenuated podocyte injury, reduced albuminuria, and diminished mesangial expansion.Furthermore, there was a reduction in the production of several pro-inflammatory cytokines, namely IL-1β,IL-6, IL-8, CCL-2, and TNF-α.These results imply that NOD1 and NOD2, and particularly NOD2, may be involved in glomerular inflammation in patients with diabetes.
3.2.NLRP3
NLRP3 plays a crucial role in the development of kidney disease,liver disease, and cancer by participating in the formation of the inflammasome complex [108-110].The inflammasome is an important component of innate immunity[102]and has the ability to sense internal molecules,such as cytokines and DAMPs,thereby facilitating the activation of diverse signaling pathways.The activation of caspases and maturation of cytokines can be triggered by inflammasomes in response to microbial invasion, further enhancing adaptive immunity.In addition to stimulating a series of caspase pathways that result in cell apoptosis,inflammasomes can activate pro-inflammatory cytokines, particularly IL-1β and IL-18.Inflammasomes were initially identified as high-molecularweight proteins composed of several domains, including the PRR,adaptor protein apoptosis-speck-like protein(ASC),and CARD[111].
As the most widely studied inflammasome, the NLRP3 inflammasome is activated by a wide range of stimuli and is instrumental in the development of many renal diseases [112].It comprises three main components: NLRP3 (a sensor molecule), ASC (an adaptor protein), and pro-caspase 1 (an effector protein) [96].Activation of the NLRP3 inflammasome requires two signals: a priming and an activation signal.In the first step, PAMP or DAMP activates NF-κB,which leads to the production of NLRP3,pro-IL-1β,and pro-IL-18.In the second signal,stimuli such as ROS,extracellular ATP,hyaluronan,uric acid, and pathogens trigger the activation of NLRP3 [113-115].Once activated, NLRP3 binds to ASC, leading to inflammasome formation through polymerization and subsequent recruitment of procaspase 1.Owing to its proteolytic activity, the inflammasome activates caspase 1, leading to the cleavage of inactivated proinflammatory cytokines and the induction of the production of activated cytokines, including IL-1β and IL-18 [95].Furthermore,activated caspase 1 cleaves gasdermin-D to produce a new fragment with a high affinity for phospholipids in the inner membrane,contributing to the formation of pores in the cellular membrane.This triggers pyroptosis[95,116],a form of programmed cell death(Fig.2).
3.2.1.NLRP3 inflammasome in DN
The pathogenesis of DN is strongly influenced by inflammasome activation.Several NLRs,such as NOD1,NOD2,and NLRP3,actively contribute to DN development.NLRP3 inflammasome is the most well-studied inflammasome complex.Activation of the NLRP3 inflammasome plays a significant role in renal inflammation observed in DN [117,118].DN is characterized by hyperglycemia,hyperlipidemia, hyperuricemia, oxidative stress, and AGE production.As a result of these factors,NLRP3 inflammasome is activated to induce the production of IL-1β and IL-18 [119].According to previous studies,both in vivo and in vitro,activation of the NLRP3 inflammasome may result from hyperglucose stimulation.NLRP3,IL-1β,and IL-18 are more prevalent in patients with DN and mouse models[120].Activation of the NLRP3 inflammasome in glomerular resident cells promotes cytokine secretion,thereby aggravating DN[119].In cultured podocytes, high glucose levels induced NLRP3 inflammasome activation, whereas blocking the activation of the NLRP3 inflammasome by NLRP3 shRNA or ASC shRNA alleviated IL-1β production and eventually attenuated podocyte injury [121].Diabetic mice with NLRP3-/-or caspase-1-/-had dramatically less albuminuria and fractional mesangial areas than wild-type diabetic mice [119].Blocking the activation of NLRP3 protected kidneys by suppressing inflammation and fibrosis in DN.Podocyte and glomerular injuries are attenuated when the NLRP3 inflammasome is blocked in DN [122].

Fig.2.The activation of nucleotide-binding oligomerization domain like receptor pyrin domain containing 3(NLRP3)inflammasome in diabetic nephropathy(DN).Activating the NLRP3 inflammasome involves two steps.The inflammasome triggers the generation of activated cytokines like interleukin(IL)-1β and IL-18.The secretion of IL-1β and IL-18 then triggers inflammatory reactions,exacerbating renal fibrosis.AGEs:advanced glycation end products;DAMPs:damage-associated molecular patterns;PAMPs:pathogen-associated molecular patterns; GSDMD: gasdermin-D; RAGE: AGEs receptor; TNF: tumor necrosis factor; ROS: reactive oxygen species; ASC: apoptosis-speck-like protein; MyD88: myeloid differentiation factor 88; TLR: Toll-like receptors.
3.2.2.Targeting NLRP3 inflammasome in DN
The NLRP3 inflammasome may be an effective target for the treatment of various inflammatory diseases.These therapies use NLRP3 or molecules upstream of NLRP3 as therapeutic targets.In db/db mice, the administration of MCC950, a small-molecule inhibitor of NLRP3, resulted in a significant reduction in active caspase 1 and IL-1β production,leading to significantly improved renal fibrosis and dysfunction.When mesangial cells were subjected to high glucose levels, the levels of activated caspase1 and IL-β were also reduced by MCC950 [123].
NLRP3 appears to be regulated by various molecules in DN.ROS,particularly mitochondrial ROS(mtROS),have been shown to play a critical role in NLRP3-dependent activation in DN [119].mtROS expression was elevated in TECs derived from DN patients and db/db mice and was positively correlated with the upregulation of NLRP3 and IL-1β expression levels.An antioxidant-MitoQ attenuated albuminuria and reduced the upregulation of NLRP3 and IL-1β in the kidneys of diabetic mice [124,125].These results suggested that the mtROS/thioredoxin-interacting protein(TXNIP)/NLRP3/IL-1β axis might be responsible for tubular damage in DN.In addition,high glucose levels triggered ROS/NLRP3 signaling partly through sweet taste receptors (STRs), which was prevented by the STR inhibitor lactisole [126].
Activation of the NLRP3 inflammasome in DN is also induced by TXNIP.TXNIP binds physiologically to the antioxidant thioredoxin(TRX).The expression is upregulated in DN.In podocytes, the binding of TXNIP to NLRP3 is a crucial mechanism for activating the NLRP3 inflammasome.Simultaneously, TXNIP activates the NLRP3 inflammasome by triggering NOX activation [121,127].Moreover,the ROS/TXNIP/NLRP3 pathway is activated by high glucose levels,resulting in the dissolution of TRX and TXNIP within mesangial cells[128].The absence of TXNIP hindered the activation of the TXNIP/NLRP3 pathway, which would have otherwise led to the activation of the NLRP3 inflammasome [129].Hyperglycemia triggers TXNIP expression, which subsequently activates gp91phox(a subunit of NADPH oxidase)and NLRP3.Furthermore,TXNIP drives the activation of NADPH oxidase, promoting subsequent activation of the NLRP3 inflammasome in podocytes [127].Both the NLRP3 inflammasome and IL-1 were suppressed when TXNIP or NADPH oxidase was inhibited in STZ-induced diabetic mice.Therefore,inhibition of TXNIP or NADPH may offer a novel approach to improve DN[130].
There are two types of purine receptors.Purinergic type 2 (P2)receptors include P2X receptors, which are ligand-gated ion channels, and P2Y receptors, which are G-protein-coupled metabolites.The kidney expresses all P2X receptor subtypes (P2X1-7)with the exception of P2X3.An additional endogenous signal,extracellular ATP, activates the NLRP3 inflammasome by stimulating P2X receptors, producing IL-1β and IL-18 [131].In DN,tubulointerstitial inflammation is advanced by high glucose levels via ATP-P2X4 signaling to activate the NLRP3 inflammasome.ATPP2X4 signaling is blocked by apyrase,which consumes extracellular ATP.P2X7R is associated with podocyte injury in DN.Evidence showed that the NLRP3 inflammasome is activated by P2X7,which causes renal inflammation.The absence of P2X7 reduced NLRP3 inflammasome activity, renal inflammation, and fibrosis [132].In addition,renal activation of the NLRP3 inflammasome is weakened by suramin (a P2 receptor antagonist), TNP-ATP (a P2X receptor inhibitor), and 5-BDBD (a P2X4 antagonist) in DN [131].
4.Chemokines
Chemokines are a class of cytokines that act as chemoattractants, directing leukocytes to areas of tissue damage or inflammation [133,134].Based on structural differences, chemokines can be classified into four subfamilies: CC, C, CXC, and CX3C[135].Chemokines are also classified into homeostatic and inflammatory subtypes based on their functions.Constant secretion of homeostatic chemokines is crucial for lymphocyte recirculation,whereas inflammatory chemokines are involved in the progression of inflammatory disorders[136].Chemokines activate intracellular signaling pathways that stimulate cellular movement and activity by interacting with G protein-coupled receptors and causing structural changes.Regions where ligands bind to chemokine receptors have been identified through mutagenesis research and creation of chimeric proteins.Studies on CCL2, CCL5, CCL9, CCL19,and CXCL10 have highlighted the significance of the N-terminal regions of these ligands in activating signaling through their respective receptors.The significance of the N termini in ligand binding has been demonstrated for various receptors such as C-C motif chemokine receptor (CCR)2, CCR3, CCR5, and CXCchemokine receptor (CXCR)1.The arrangement of the intricate region revealed that the N-terminus of CXCR1 attaches to CXCL8 in a prolonged manner, potentially aligning the N-terminus of the chemokine into the helical bundle.In addition to the N-terminus,extracellular loops of receptors participate in ligand attachment[137].Various cell types express chemokines and their receptors[138].For instance, CCL chemokines interact with CCR1,CCR2,and CCR5 to attract most monocytes.CXCR1 and CXCR2 are receptors targeted by CXCL chemokines, which activate neutrophils.Chemokines that bind to CXCR3, CXCR6, CCR5, and CX3CR1 recruit Thelper1 cells and natural killer cells to stimulate inflammatory responses mediated by type 1 cytokine (IL-2, INF-γ).Upon stimulation, the renal endothelial cells, podocytes, mesangial cells, TECs,and fibroblasts produce chemokines [139,140].
The migration of inflammatory cells to the kidneys is significantly influenced by chemokines, and DN is strongly correlated with their activity.Studies on diabetes in humans and animals have demonstrated an increase in the infiltration of monocytes and macrophages into the renal glomeruli and interstitium[13,141,142].Chemokines and their respective receptors have been proven to participate in this process and contribute to kidney damage in patients with diabetes [139,143].Various inflammatory chemokines are significant contributors to renal injury in diabetes, and these chemokines are upregulated due to metabolic and hemodynamic changes caused by diabetes.Mesenchymal stem cells release chemokines in DN under inflammatory conditions [144].Animal models of DN have shown an increase in the levels of various chemokines in the glomeruli and proximal tubules,including CCL2,CCL20, CXCL5, CXCL7, and CXCL12 [145].
4.1.MCP-1/CCL2 in DN
A family of CC chemotactic chemokines called CCL2 or MCP-1,is secreted by monocytes and renal resident cells and binds to CCR2.Numerous inflammation-related disorders, including DN, exhibit increased CCL2 expression.It is primarily CCL2 that governs monocyte recruitment to inflammatory sites.By triggering CCR2,CCL2 promotes monocyte release from the bone marrow and recruits monocytes to the inflammatory areas.The CCL2/CCR2 pathway contributes significantly to the pro-inflammatory actions in DN[146,147].In DN,CCL2 is associated with renal inflammation and tubular injury[148].Renal inflammation,tubular atrophy,and interstitial fibrosis may all be mediated by CCL2,which is produced by intrinsic renal cells, such as podocytes, mesangial cells, and tubular epithelial cells [149,150].
In kidney tissue samples obtained from DN patients, CCL2 was up-regulated.According to clinical findings, patients with DN experience a consistent increase in urinary CCL2 levels, especially those at an advanced stage of the complication.In the serum of patients with DN, CCL2 concentration is associated with inflammatory processes and renal fibrosis [151].Urinary levels of CCL2 serve as crucial indicators for predicting the swift deterioration of renal function in individuals with DN [152,153].Furthermore, in T2DM patients with preserved renal function,the urinary CCL2-tocreatinine ratio was related to persistent renal failure [154].According to many experiments on diabetes,diabetic glomeruli at an early stage of the disease revealed that CCL2 was overexpressed[142].Overexpression of CCL2 during diabetes development has also been linked to macrophage infiltration [149,155].Most CCL2 overexpression in the glomeruli may be caused by podocytes[156].In contrast, tubular cells in experimental diabetes and in patients with DN were subject to the upregulation of CCL2 [149,157].The increase in CCL2 and interstitial macrophages in the renal tubules was consistent with hyperglycemia during experimental diabetes.In vitro studies, the expression of CCL2 in human renal TECs was negatively regulated by miR-374a [158].
The recruitment and differentiation of inflammatory cells and macrophages are modulated by CCL2.Macrophages are activated by high glucose levels via TGF-β/MAPK and NF-κB pathways, thereby polarizing macrophages to pro-inflammatory phenotype and speeding up DN [159].In podocytes, the upregulation of TLR4 and CCL2 is mediated by high glucose levels through NF-κB-dependent signaling [66].Inflammatory cytokines and chemokines, including CCL2 and TNF-α, are stimulated by the activation of TLRs and thus play crucial roles in DN development [80].Elevated glucose levels increase the expression of CCL2 and facilitate the activation of NFκB-p65,IκB kinase(IKK)β,MAPK,and the nuclear translocation of p65 in podocytes.Additionally, high glucose levels-induced podocyte damage upregulated the receptor activator for NF-κB[160].In mouse proximal tubular cells, the ROS/Ras/NF-kB signaling pathway increased the dose- and time-dependent production of CCL2 by AT1R and AT2R [161].Further, TGF-β1 is capable of activating nicotinamide adenine dinucleotide phosphate (NAD(P)H)oxidase/p67phox-dependent mechanism to raise the levels of CCL2 mRNA and protein [162].Persistent hyperglycemia increases the generation of AGEs,which,in turn,increases the expression of CCL2 in mesangial cells.Exposure of mesangial cells to AGEs resulted in the overexpression of CCL2, possibly due to AGEs binding to the AGEs receptor (RAGE) [163,164].Studies conducted in vitro demonstrated that TGF-β1 enhanced the expression of CCL2 by activation of ERK and p38 MAPK and formation of ROS in murine mesangial cells [165].CCL2 promotes the generation of TGF-β1,indicating a positive feedback loop between TGF-β1 and CCL2[166].Additionally, the CCL2 pathway is also triggered by connective tissue growth factor, a prosclerotic cytokine that functions downstream of TGF-β1 [167].
4.2.Targeting CCL2/CCR2 system
According to several recent studies, the CCL2/CCR2 system has been specifically targeted by novel strategies.RAS inhibitors and statins have been proven to lower the renal expression of CCL2 and the recruitment of macrophages among the currently approved drugs for patients with DN.In STZ-induced diabetic rats,the use of either an ACE inhibitor or an AT1 receptor antagonist for RAS inhibition resulted in decreased renal CCL2 expression and macrophage infiltration [168].Lisinopril treatment for T2DM individuals with DN resulted in improved albuminuria and creatinine clearance, as well as decreased urinary concentrations of CCL2 [169].Among these patients, a strong correlation was observed between improved proteinuria and reduced urinary CCL2 levels.Baricitinib,a januaryus kinase (JAK)1/JAK2 inhibitor, and emapticap pegol, a CCL2 inhibitor, are promising DN drugs for clinical trials [170].During a phase II clinical trial, T2DM patients at high risk of progressive DN showed a lower urinary albumin-creatinine ratio when baricitinib downregulated urinary CCL2 expression [171].Furthermore, several CCR2 antagonists have been found to have positive effects in diabetes models.RS102895 and RS504393 significantly ameliorated insulin resistance and decreased the concentration of urinary albumin and infiltration of macrophages in diabetic mice[172,173].CCX140-B,a CCR2 inhibitor,also has beneficial effects on DN [174].Inhibitors of the CCL2/CCR2 pathway may be promising treatments for improving renal outcomes in patients with DN;however, further studies are required[170].
5.The complement system
The complement system is one of the most crucial components of the innate immune system, facilitating the clearance of pathogens and damaged cells from the body, while also promoting inflammation.Dysregulation of the complement system has been implicated in various diseases and kidney damage [175-177].
There are three main ways to activate the complement cascade:the lectin, classical, and alternative pathways [178] (Fig.3).The lectin pathway is typically activated by the mannose-binding lectins(MBL).A set of macromolecules,known as pattern recognition molecules (PRMs), including H-ficolin, L-ficolin, and M-ficolin, can trigger the activation of this pathway[179-181].Activation of MBLassociated serine proteases (MASPs) then cleaves C2 and C4 [181].Immune complexes such as IgG or IgM bind to the C1q domain of complement C1 to activate the classical pathway.The non-covalent complex, C1, comprises three distinct proteins: C1q, C1r, and C1s.However,both the lectin and alternative pathways can be activated in an antibody-independent manner.The alternative pathway is continuously activated at a low level called tickover and is further amplified after exposure to a variety of proteins and carbohydrate structures on microbes and other foreign surfaces[182].To enhance the complement cascade, the C3b fragment generated through all complement pathways can attach to factor B and, with the assistance of factor D,create an additional C3bBb C3 convertase.When C3b fragments bind to C3 convertases, they form C5 convertases that split C5 into C5a and C5b fragments.C5b subsequently associates with C6, C7, C8, and C9 to establish a membrane attack complex(MAC)[179].The MAC creates a channel in the membrane that results in bacterial lysis and cell death [183].In addition, the C3a and C5a fragments (known as anaphylatoxins) function as effectors by binding to and activating their G-protein-coupled receptors C3aR and C5aR1.This results in the release of cytokines and chemokines and the activation of macrophages,antigen-presenting cells, and T cells [184].
5.1.The complement system in DN
Research has shown that most of the active components of the complement cascade can be synthesized by the kidneys [185].Increasing evidence suggests that DN is influenced by the complement system.The pathogenesis of DN is linked to complement activation.The complement system is thought to contribute to DN pathogenesis via two primary mechanisms [178].First,hyperglycemia-induced expression of glycated proteins on cell surfaces leads to activation of the lectin pathway through binding of PRMs [186].Second, elevated blood glucose levels lead to the glycation of proteins that regulate the complement system,resulting in the impairment of their regulatory capabilities.Thus,the overactivation of complement pathways may aid complement auto-attacks [187].
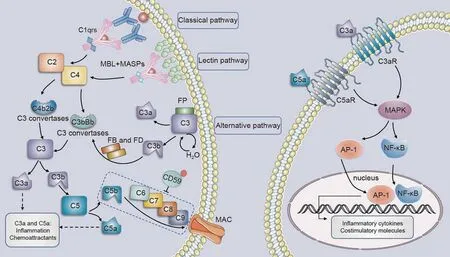
Fig.3.Potential activation of complement in diabetic nephropathy(DN).MASPs:MBL-associated serine proteases;MBL: mannose-binding lectin; FB:factor B; FD:factor D;MAC:membrane attack complex; MAPK: multiple mitogen-activated protein kinase; AP-1: activator protein-1; NF-κB: nuclear factor-kappa-B.
Proteomic analysis of kidney biopsies of glomeruli from patients with DN revealed that the complement protein was upregulated[188].The concentrations of MBL, C3a, C5a, and soluble MAC are markedly elevated in both blood and urine samples of individuals with DN [189].Most of these factors that have been linked to the intrarenal generation of MAC are overexpressed in the kidneys of diabetic rats [190].Several clinical studies have shown that the lectin pathway is hyperactivated in DN patients.T1DM patients with DN exhibited significantly higher MBL levels than those with normoalbuminuria [191].Another study reported that persistent microalbuminuria or macroalbuminuria was associated with higher serum MBL levels in the early stages of T1DM [192].Moreover, an association between genotypes linked to elevated circulating levels of MBL and the onset of DN as well as increased mortality in T1DM patients has been identified[191].Individuals with T1DM who had high MBL genotypes were found to have a 50% increased risk of progression to kidney disease [193], as well as significantly higher mortality rates during a ten-year follow-up period[194].Similar to the clinical trials cited earlier, several laboratory experiments have provided evidence for a causal link between MBL and DN.In STZinduced diabetes models, the kidneys of MBL-deficient mice exhibited lower weights, decreased levels of albumin in urine, and reduced fibrosis compared to wild-type diabetic animals [195].Additional animal research utilizing this model revealed that there was not only an increase in endogenous MBL accumulation in glomerular tissue but also an increase in MBL half-life after diabetes onset [196,197].Altered glomerular cell surfaces caused by hyperglycemia are expected to be identified by MBL,which may aid in the pathophysiology of DN and the activation of autoreactivity.
Studies have suggested that C3 and C5 play important roles in DN.In renal biopsy specimens obtained from individuals diagnosed with DN, transcriptome and immunohistochemical analyses revealed a six-fold increase in glomerular C3 levels [198].Furthermore, patients with macroalbuminuria and T2DM have markedly elevated plasma levels of C3 compared to those with normal albuminuria [199].Both non-obese diabetic mouse [200]and OVE26 diabetic mouse [201] models of T1DM demonstrate elevated expression of C3 in their kidneys.Similar findings were found in KK mice, a model of T2DM in which C3 was deposited in the glomeruli and glomerular capillaries.Signaling via C3a and its receptor in podocytes leads to cytoskeletal remodeling and impaired mitochondrial function in genetically obese(ob/ob)mice,resulting in proteinuria and podocyte loss[202].The renal tubules showed higher levels of C5a in patients with DN, and there was a clear correlation between C5a levels and renal disease progression in these patients.Another study demonstrated that tubular deposition of C5a correlated with the severity of DN.In a murine diabetic model, blocking C5a/C5aR signaling improved interstitial fibrosis[203].Changes in mitochondrial agility were observed in STZinduced diabetic mice, which were restored following the suppression of C5aR.Therefore, targeting C5aR and C3aR is an appealing option for protecting diabetic kidneys.
Additionally, it appears that diabetes is associated with increased levels of glycation,which can result in the dysfunction or inactivation of proteins that regulate the complement system.CD59, for instance, can inhibit MAC to prevent the harmful effects of complement overactivation [187,195].Inactivation of CD59 due to glycation and activation of complement signaling caused by hyperglycemia have been proposed to enhance MAC deposition[178].In a small cohort of patients with T2DM,CD59 was identified as one of the most downregulated genes in glomeruli [204].Furthermore, the increased secretion of urinary CD59 contributes to a reduced risk of ESRD [205].
5.2.Targeting the complement system
Significant efforts have been made to develop inhibitors that target complementary pathways.As a result of C3aR antagonism,both proteinuria and decline in renal function were prevented in the ob/ob murine model of DN, while the loss of podocytes was reduced [206].SB290157, a C3aR antagonist, lowers albuminuria and improves renal fibrosis in a rat T2DM model.Under hyperglycemic conditions,SB290157 treatment effectively suppresses the expression of inflammatory and fibrotic markers in cultured human glomerular endothelial cells [207].K-76 COONa, a C5aR inhibitor,similarly decreases glomerular lesions and DN severity in rats[208].In db/db mice,treatment with NOX-D21,an inhibitor of C5a,resulted in a decrease in glomerulosclerosis and tubulointerstitial damage, as well as a reduction in lipid deposition in renal tissue[209].Blocking of C3a and C5a receptors improved endothelial-tomyofibroblast transition by the Wnt/β-catenin signaling pathway in the T1DM model [207].Numerous preclinical studies have demonstrated the efficacy of C3aR and C5aR1 inhibitors in protecting the kidneys from DN damage.Nevertheless, additional research is necessary to fully understand the involvement of C3a/C3aR and C5a/C5aR signaling in the pathogenesis of inflammation and fibrosis in DN.Further investigation is required to examine the functions of additional elements of the complement system,including complement regulators and upstream C3aR, in the progression of DN.Currently, it is unclear how best to target the complement system to prevent the development of DN.It is crucial to consider the possible negative consequences of prolonged inhibition of the complement system in chronic illnesses,especially the heightened vulnerability to infections.
6.Conclusions
The pathogenesis of DN is significantly influenced by inflammation, and anti-inflammatory processes may have a significant impact on the protective effects on the kidneys.Tissue damage activates innate immunity through the recognition of DAMPs by various PRRs during hyperglycemia, inducing a series of inflammatory reactions.Persistent inflammation caused by long-term renal damage may lead to DN development.Various proinflammatory pathways are involved in the progression of DN.Several studies targeting the innate immune pathways in DN have demonstrated promising outcomes.Considering the critical role of inflammatory pathways, a combined treatment approach that includes both anti-inflammatory and antidiabetic medications may offer better protection against DN.However, clinical trials on anti-inflammatory treatments are only beginning.Patients may become more susceptible to infection in the absence of specific anti-inflammatory treatments.Additionally, TLR suppression in patients with DN may increase the risk of tumorigenesis and the recurrence of malignancies.Therefore, the regulation of innate immune pathways and the development of more specific and less toxic therapies for DN need to be further explored in the future.
CRediT author statement
Min Yang:Writing - Original draft preparation, Reviewing and Editing, Funding acquisition;Chun Zhang:Resources, Writing -Reviewing and Editing, Supervision, Funding acquisition.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant Nos.: 82100801, 81974096,81770711, 81974097, and 81961138007).
 Journal of Pharmaceutical Analysis2024年1期
Journal of Pharmaceutical Analysis2024年1期
- Journal of Pharmaceutical Analysis的其它文章
- Lipid metabolism analysis in esophageal cancer and associated drug discovery
- Push forward LC-MS-based therapeutic drug monitoring and pharmacometabolomics for anti-tuberculosis precision dosing and comprehensive clinical management
- Metformin: A promising clinical therapeutical approach for BPH treatment via inhibiting dysregulated steroid hormones-induced prostatic epithelial cells proliferation
- Epimedin B exhibits pigmentation by increasing tyrosinase family proteins expression, activity, and stability
- Hydralazine represses Fpn ubiquitination to rescue injured neurons via competitive binding to UBA52
- Distinct molecular targets of ProEGCG from EGCG and superior inhibition of angiogenesis signaling pathways for treatment of endometriosis