
Aβ1-42诱导的阿尔茨海默病细胞模型中神经损伤及凋亡的机制
2024-03-22 12:12王昱驰马宏玉王振宇张少轩张育泰高宇航孙嘉伟石河子大学医学院基础医学系病理生理学教研室石河子832002通讯作者mailjiaweisun01163com
山西医科大学学报 2024年2期
王昱驰,马宏玉,王振宇,张少轩,张育泰,高宇航,孙嘉伟(石河子大学医学院基础医学系病理生理学教研室,石河子 832002;通讯作者,E-mail:jiaweisun01@163.com)
AD是老龄化人口中普遍存在的一种进行性神经系统退行性疾病,主要以脑皮质与海马神经元丢失、β-淀粉样蛋白(Aβ)沉积和Tau蛋白磷酸化(p-Tau)引起的神经纤维缠结为特征,其临床表现为进行性认知功能障碍和记忆力损害[3-5]。AD可导致神经元细胞的广泛凋亡,但其具体致病机制至今仍不明确[6]。已知细胞凋亡中最主要的病理性生化事件为半胱天冬酶(cysteiny laspartate specific proteinase,CASP)级联反应的激活,且半胱氨酸酶-3(Caspase-3)是细胞凋亡过程中最主要的终末剪切酶,研究表明在AD患者的样本中检测到其水平的改变[7]。早期AD中常见的病理变化为神经元突触素(synapsin,SYP)、维持微管稳定的蛋白(microtubule associated protein 2,MAP-2)的降低导致突触处于不稳定状态甚至损伤,以及淀粉样蛋白(amyloid β,Aβ)不受控制的寡聚化和聚集造成的神经元细胞凋亡[8]。故我们选择Caspase-3、SYP、MAP-2作为鉴别AD神经元的蛋白指标。此外,神经突蛋白1(Neuritin 1,NRN1)是一种与神经发育和神经可塑性密切相关的营养因子,对损伤神经元具有一定的修复作用[9],其在神经元中的表达具有一定的积极意义。
Aβ来自于其前体淀粉样蛋白前体蛋白(amyloid precursor protein,APP)第672~711残基裂解片段。有研究表明,Aβ1-42是AD患者脑中主要的淀粉样蛋白沉积物,且Aβ1-42是Aβ中传播性最强的亚型[10,11]。据报道,过多的Aβ可直接或间接通过影响线粒体的结构或者功能进而诱发氧化应激、激活凋亡信号通路等级联反应,导致大量的神经元细胞损伤[12]。因此,结合Aβ的作用结果以及AD的病理特征,我们选择Aβ1-42来诱导AD细胞模型的建立。
目前治疗AD药物的研发进展缓慢,AD预防和治疗依然是相关研究领域的热点[2]。已有的细胞模型缺乏对凋亡机制的预测和研究,因此建立适宜的AD细胞模型,有利于进一步研究AD发病机制及其预防和治疗方法。本实验拟通过Aβ1-42诱导小鼠海马神经元细胞HT22建立AD细胞模型,并通过生物信息学分析进一步明确Aβ1-42导致细胞凋亡的相关信号通路。
1 材料与方法
1.1 材料与试剂
Aβ1-42寡聚肽购自上海强耀科技有限公司;10%FBS和0.25%Trypsin-EDTA购自美国Thermo Fisher公司;DMEM培养基购自美国HyClone公司;0.01 mol/L PBS购自美国Sangon公司;Triton X-100购自加拿大Biosharp公司;BSA购自德国VETEC公司;PVDF膜购自美国Immobilon公司;RIPA裂解液、10%SDS、SDS-PAGE电泳液、CCK-8试剂盒、BCA蛋白浓度测定试剂盒均购自上海碧云天生物技术有限公司;cleaved-Caspase-3抗体、Caspase-3抗体、ERK抗体、p-ERK抗体均购自美国CST公司;MAP-2抗体、SYP抗体和NRN1抗体购自英国abcam公司;β-actin抗体以及山羊抗兔IgG、山羊抗小鼠IgG二抗均购自北京中杉金桥生物技术有限公司。
1.2 设备
生化培养箱购自上海博迅医疗生物仪器股份有限公司;荧光倒置显微镜购自德国ZEISS公司;多功能酶标仪与高速离心机购自美国Thermo Fisher公司;免疫印迹反式半干转仪购自美国Bio-rad公司;全自动化学成像仪购自上海天能生命科学有限公司。
1.3 方法
1.3.1 细胞培养及实验分组 HT22细胞株由黄瑾教授课题组惠赠,使用含10%FBS的DMEM培养基在37 ℃,5%CO2的细胞培养箱中培养,细胞每隔2 d传一代,传代前1 d半量换液,定期观察细胞形态及生长、贴壁情况。将HT22细胞悬液转移至96孔细胞培养板中(100 μL/孔,约5 000个/孔),在细胞培养箱中孵育,待细胞贴壁;使用0.01 mol/L PBS(pH=7.4)溶解Aβ1-42粉末,配制不同浓度的Aβ1-42溶液(0,0.625,1.25,2.5,5,10,20,40 μmol/L),置于-80 ℃冻存,使用前置于37 ℃下老化7 d。分别使用各浓度Aβ1-42溶液干预处于生长对数期的细胞,其中Aβ1-42浓度为0 μmol/L的细胞中仅添加同体积PBS,以便与其他组别形成对照。每个浓度设置5个复孔,2 d后观察细胞形态、检测细胞存活率。每组实验重复3次,确定建立AD细胞模型的最适浓度;随后使用最适浓度干预HT22细胞,分别在6,12,24,48 h后观察细胞形态、检测细胞存活率。每组实验重复3次,确定建立AD细胞模型的最适时间。
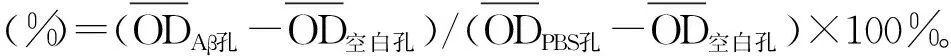
1.3.3 AD细胞模型的特征性蛋白鉴定 为明确Aβ1-42干预HT22细胞建立AD细胞模型的有效性,在Aβ1-42干预HT22细胞建立AD细胞模型的最适条件下,将细胞分为AD组(Aβ1-42+PBS)和NC组(PBS)。AD组取已用PBS配制好的20 μmol/L Aβ1-42溶液,NC组取同体积PBS溶液,分别干预处于生长对数期的细胞24 h。提取各组细胞蛋白,使用BCA试剂盒测定各组蛋白浓度,Western blot检测cleaved-Capase-3、SYP、NRN1、MAP-2、ERK蛋白表达情况,免疫荧光检测MAP-2分布情况。一抗分别为anti-cleaved-Caspase-3(1∶1 000)、anti-Caspase-3(1∶1 000)、anti-SYP(1∶5 000)、anti-MAP-2(1∶1 000)、anti-NRN1(1∶1 000)、anti-ERK(1∶1 000)、anti-p-ERK(1∶2 000),4 ℃孵育过夜;TBST洗膜4次,每次8 min,室温下孵育二抗(1∶5 000)2 h,TBST洗膜4次,每次8 min,室温孵育2 h,使用全自动化学成像仪曝光,以anti-β-actin(1∶1 000)作为内参。
样品配制:0.2 mL臭牡丹粗提物溶液(5.0 mg/mL)与0.2 mL DPPH甲醇溶液(25.0 mg/mL)混合均匀,37 °C下避光孵育30 min,直接进行HPLC-QTOF-MS/MS检测.等体积甲醇替代DPPH自由基溶液作为空白对照组.
制作AD组和NC组细胞爬片,使用anti-MAP-2(1∶200)一抗4 ℃孵育过夜;孵育后第2天,室温静置30 min,使用PBS清洗3次,每次5 min;滴加二抗(FITC Goat anti-Rabbit IgG,1∶100),37 ℃孵育30 min。PBS洗3次,每次5 min。使用含DAPI的封片剂封片,避光阴干,荧光倒置镜下观察各组细胞MAP-2荧光分布情况。从滴加二抗开始,全程避光。
1.3.4 生物信息学分析 为探索Aβ、ERK与凋亡三者间可能存在的相关性,在Genecards上检索了Aβ、ERK与Caspase-3相关基因。同时,在美国国家生物信息技术中心(National Center for Biotechnology Information,NCBI)网站GEO Data Sets数据库中(https://www.Ncbi.nlm.nih.gov/gds)以阿尔茨海默病和海马体为指标进行检索,排除表观遗传学的影响,获得了在AD模型海马体CA1中表达的基因数据集(GSE28146)。之后,筛选出数据集中AD样本(n=22),通过boxplot进行箱线图绘制使得数据标准化,使用Spearman相关分析来描述不考虑分布情况的相关基因变量之间的相关性,并通过R软件包pheatmap进行多基因相关性热图的展示,P<0.05被认为差异具有统计学意义。
2 结果
2.1 确定Aβ1-42建立AD细胞模型的最适浓度
2.1.1 不同浓度下细胞形态学变化 倒置显微镜下观察细胞形态,结果显示:正常HT22细胞(Aβ1-42浓度为0时),细胞伸展良好、细胞透亮,神经突明显;随着Aβ1-42浓度梯度的增加,细胞形态变化明显,体积变小,核固缩、细胞碎片明显增多,存活细胞数量明显减少(见图1)。
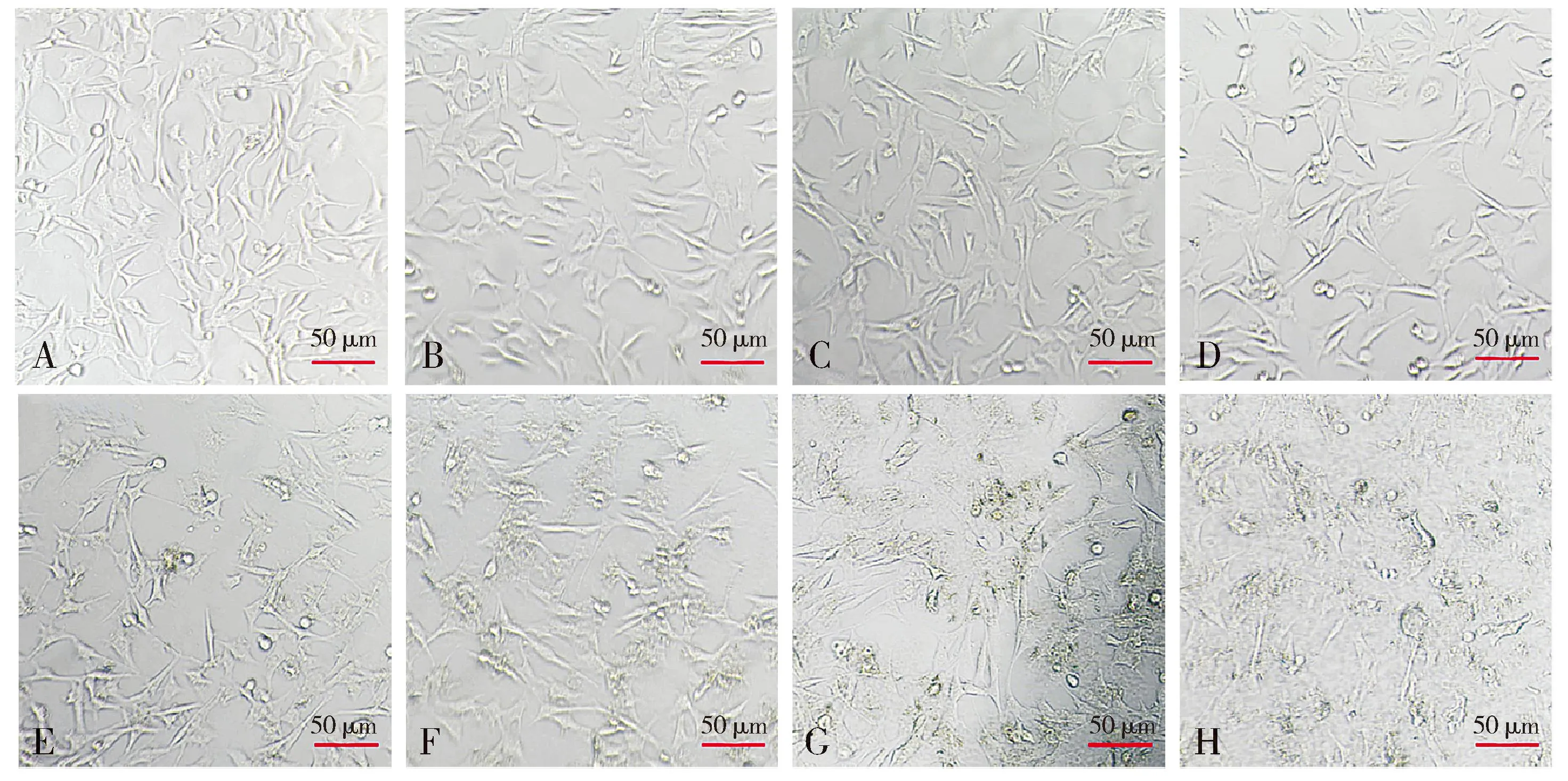
注:A-H.Aβ1-42浓度分别为0,0.625,1.25,2.5,5,10,20,40 μmol/L。图1 不同浓度Aβ1-42干预HT22细胞的形态观察Figure 1 Morphological changes of HT22 cells after intervened with different concentrations of Aβ1-42
2.1.2 不同浓度下细胞存活率变化 CCK-8结果显示,随着Aβ1-42浓度的增加,细胞活性逐渐降低;与正常HT22细胞相比,20 μmol/L Aβ1-42作用后,HT22细胞存活率降低,差异具有统计学意义(P<0.000 1,见图2),且部分存活细胞仍能保持正常细胞的形态。结合形态学观察和存活率检测,选择20 μmol/L作为Aβ1-42干预HT22细胞建立AD细胞模型的最适浓度。
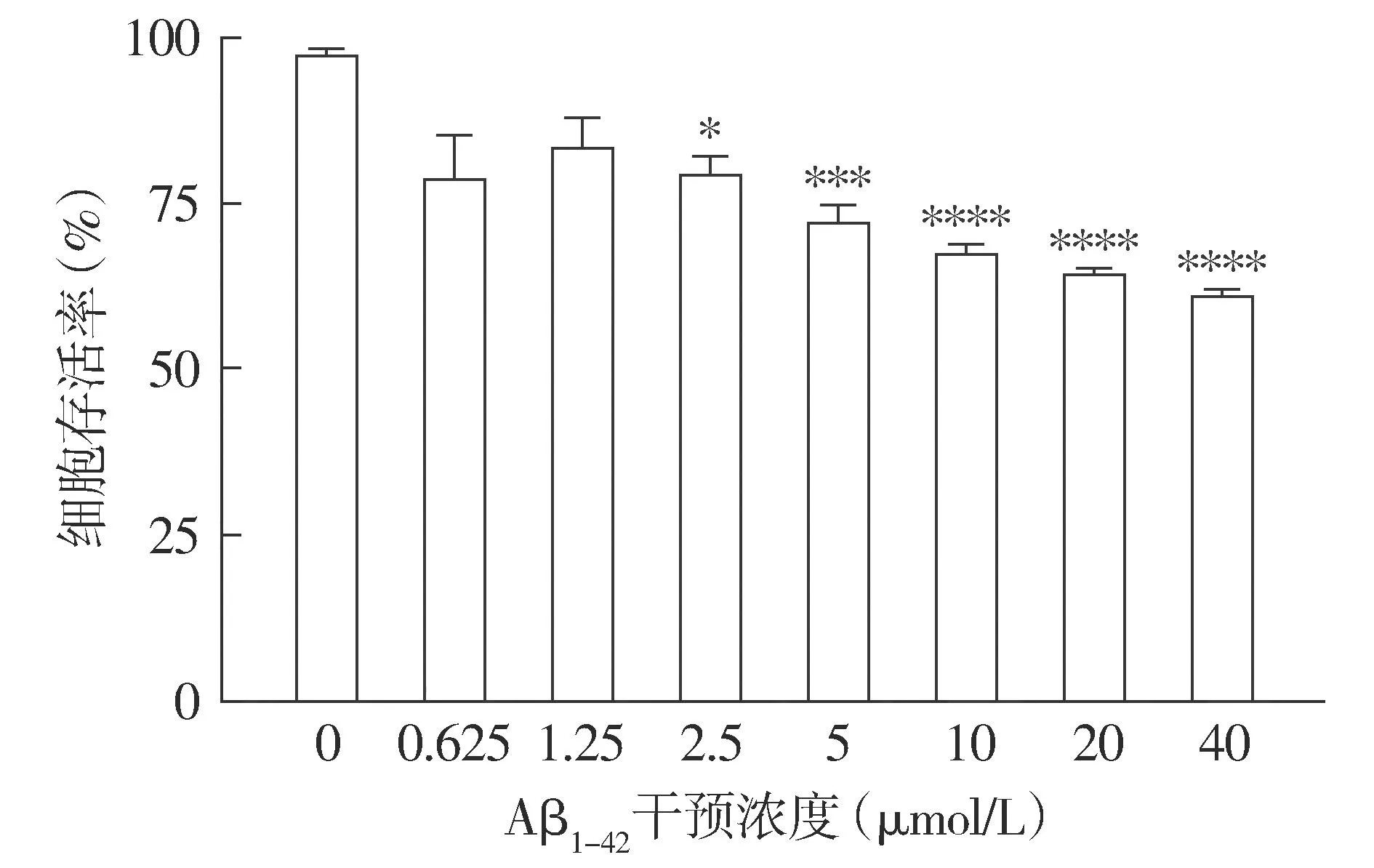
注:与0 μmol/L相比较,*P<0.05,***P<0.001,****P<0.000 1。图2 不同浓度Aβ1-42干预后HT22细胞存活率的变化Figure 2 Viability of HT22 cells after intervened with different concentrations of Aβ1-42
2.2 确定Aβ1-42建立AD细胞模型的最适时间
2.2.1 不同干预时间后细胞形态学变化 根据上述实验,我们使用Aβ1-42的最适浓度20 μmol/L干预HT22细胞,分别在6,12,24,48 h观察各组细胞形态学变化。结果显示:随着干预时间的延长,细胞胞体收缩、变形,细胞膜完整性破坏,细胞外基质杂乱不清,可见较多凋亡、坏死细胞(见图3)。

图3 不同时间Aβ1-42干预HT22细胞的形态观察Figure 3 Morphological changes of HT22 cells after Aβ1-42 intervention for different time
2.2.2 不同干预时间后细胞存活率变化 CCK-8结果显示,随着干预时间延长,Aβ1-42对细胞的毒性作用明显增加,细胞活性呈整体降低趋势(见图4);与6 h相比,干预24 h时,细胞存活率降低,差异有统计学意义(P<0.001,见图4),且部分存活细胞仍能保持正常细胞的形态。结合形态学观察和存活率检测,选择24 h作为Aβ1-42干预HT22细胞建立AD细胞模型的最适时间。
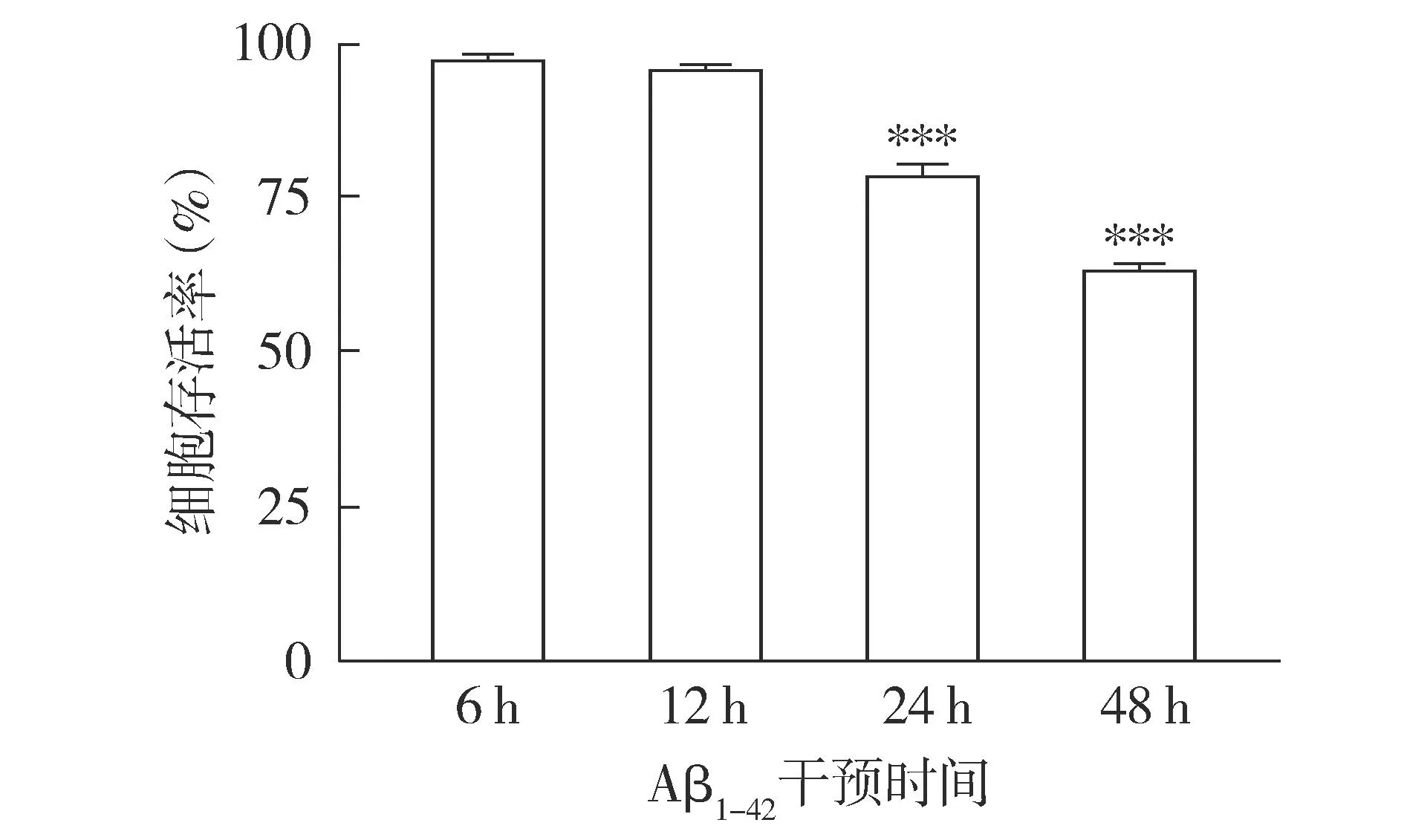
注:与6 h相比较,***P<0.001。图4 20 μmol/L Aβ1-42干预不同时间后HT22细胞存活率变化Figure 4 Viability of HT22 cells after intervention with 20 μmol/L Aβ1-42 for different time
2.3 Aβ1-42干预HT22细胞建立AD细胞模型的特征性蛋白鉴定
2.3.1 AD细胞模型的凋亡蛋白及神经营养因子检测 Western blot结果显示,与NC组相比,AD组中cleaved-Caspase-3的表达明显增多,且cleaved-Caspase-3/Caspase-3比值升高(P<0.01,见图5);AD组中NRN1蛋白的表达降低(P<0.000 1,见图5)。
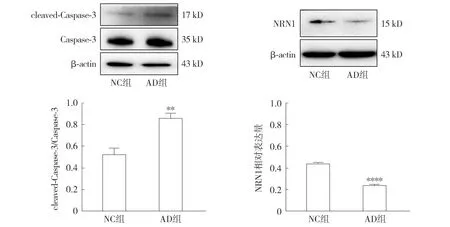
注:与NC组相比较,**P<0.01,****P<0.000 1。图5 Western blot检测Aβ1-42对HT22细胞凋亡及神经营养相关蛋白表达的影响Figure 5 Effect of Aβ1-42 on expressions of apoptosis- and neurotrophic-related proteins in HT22 cells detected by Western blot
2.3.2 AD细胞模型的神经突触生长检测 Western blot结果显示,与NC组相比,AD组SYP蛋白与MAP-2蛋白的表达降低(P<0.01,见图6)。免疫荧光检测各组MAP-2在HT22细胞中的表达及分布情况,结果显示:AD组细胞核周围的MAP-2蛋白含量明显减少(见图7)。
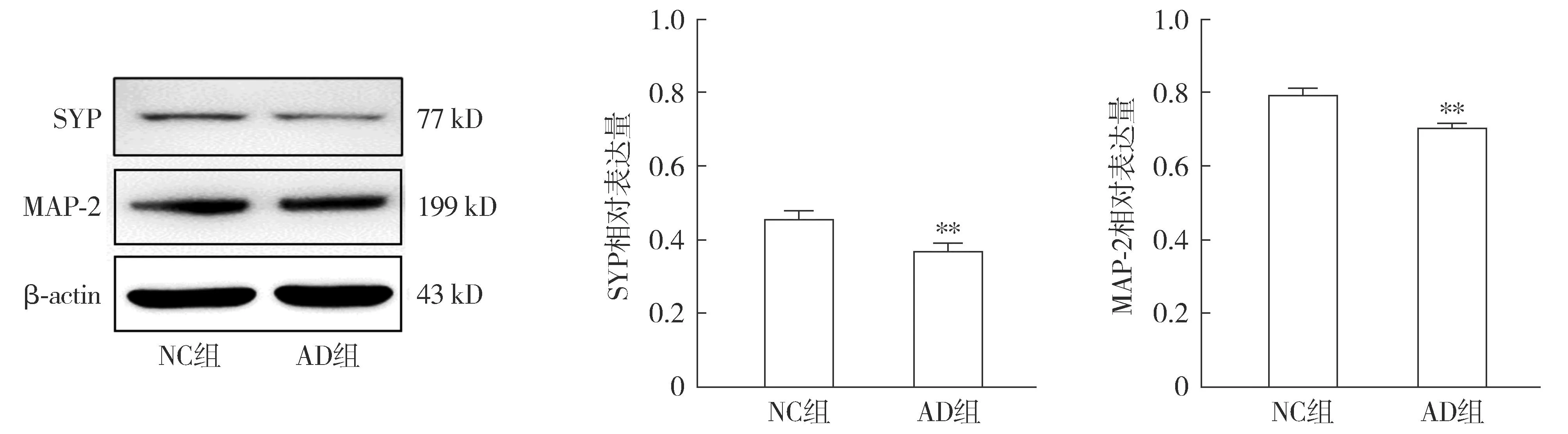
注:与NC组相比较,**P<0.01。图6 Western blot检测Aβ1-42对HT22细胞神经突触生长相关蛋白表达的影响Figure 6 Effects of Aβ1-42 on expressions of synaptic growth-related proteins in HT22 cells detected by Western blot
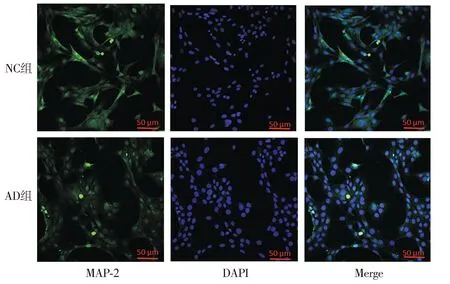
注:MAP-2为绿色荧光(FITC),细胞核为蓝色荧光(DAPI)。图7 AD细胞模型中MAP-2免疫荧光结果Figure 7 Immunofluorescence results of MAP-2 in AD cell models
2.4 AD模型相关信号通路的生物信息学分析及鉴定
2.4.1 AD模型相关信号通路的生物信息学分析 Genecards检索结果示Aβ和ERK1/2蛋白的结构基因为APP、MAPK3、MAPK1;Swiss-Prot数据库结果示Caspase-8,9,10参与凋亡通路的激活,其结构基因分别为CASP8、CASP9、CASP10。相关性分析结果显示:22个AD样本基因表达量基本相同,可认为样本同质(见图8A);MAPK3(ERK1)与APP呈明显正相关(r=0.634,P<0.01),与CASP9(Caspase-3重要的激动剂)呈正相关(r=0.513,P<0.05,见图8B)。
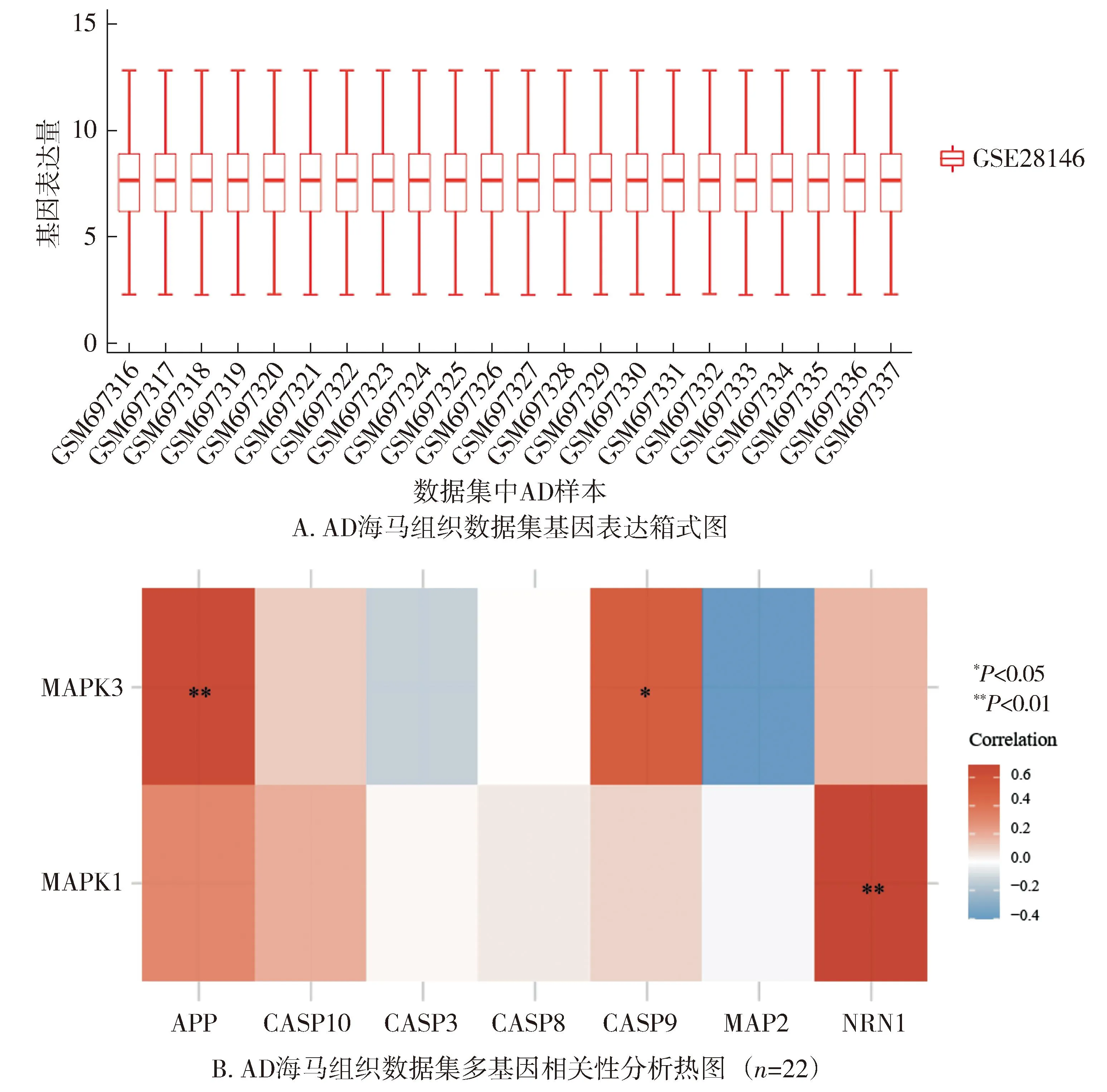
图8 AD模型相关信号通路的生物信息学分析Figure 8 Bioinformatics analysis of signaling pathways associated with AD models
2.4.2 AD细胞模型中ERK信号通路变化的检测 Western blot结果显示,与NC组相比,AD组中p-ERK/ERK比值升高,差异具有统计学意义(P<0.05,见图9)。表明本文建立的AD细胞模型中ERK激活增多,提示ERK信号通路的激活可能与AD进展(Aβ表达增加)相关。
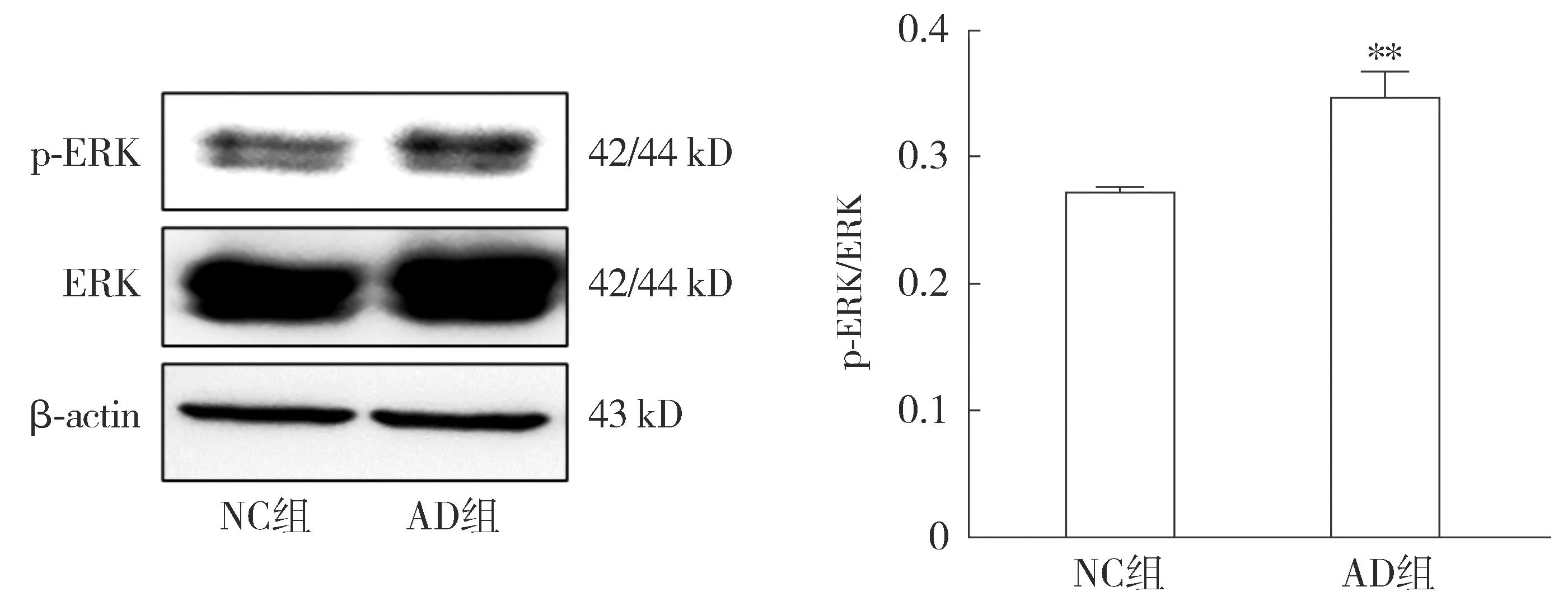
注:与NC组相比较,**P<0.01。图9 Western blot检测Aβ1-42对HT22细胞ERK信号通路相关蛋白表达的影响Figure 9 Effects of Aβ1-42 on the expressions of ERK signaling pathway-related proteins in HT22 cells detected by Western blot
3 讨论
AD是一种常见的神经系统退行性疾病,其核心表现为脑内淀粉样斑块堆积和MAPT/tau神经纤维缠结。Aβ清除失调对于其在大脑内的积聚和斑块的形成具有重要意义[13]。目前的研究仍然无法明确AD疾病发病的确切机制,它的诊断治疗和干预措施也受到限制[14]。关于AD的发病机制,目前提出了淀粉样蛋白学说、细胞凋亡学说、氧化应激学说、钙超载学说、胆碱能学说等多种学说[15,16]。研究表明,AD的发生发展多与神经元丢失、神经元凋亡密切相关[17]。为进一步探索其发生机制,我们建立了适宜的AD细胞模型。
本实验采用不同浓度梯度的Aβ1-42干预HT22细胞,在一定范围内,Aβ1-42浓度和干预时间均与HT22细胞活性整体呈负相关,较高浓度和较长干预时间的Aβ1-42能够更好地诱导HT22细胞分化成AD样细胞。因此,根据细胞存活率以及镜下观察结果得出,当Aβ1-42浓度为20 μmol/L,干预时间为24 h时,能够保证HT22细胞在较高存活率的前提下,在细胞水平上表现出明显的早期AD样特征:神经元结构受损、突触可塑性降低、和部分神经元凋亡。
为确保模型的有效性,本实验通过Western blot和免疫荧光来分析AD组和NC组在细胞凋亡相关蛋白Caspase-3,神经突触生长相关蛋白SYP、MAP-2,神经营养因子NRN1的表达差异。结果显示:与NC组相比,AD组凋亡活化指标cleaved-Caspase-3蛋白表达升高,神经营养因子NRN1表达降低,神经元结构相关指标SYP、MAP-2蛋白表达降低。上述结果表明AD组细胞凋亡增多、神经元结构受损、突触稳定性下降。这与AD早期病理变化特征相一致,提示Aβ1-42对HT22细胞具有明显的毒性作用。以上实验证明我们所建立的AD细胞在蛋白水平上具有较典型的AD样特征,即可认为Aβ1-42干预浓度和时间分别为20 μmol/L和24 h,为较理想的AD建模条件。
大量证据表明,神经元的丢失与多种信号通路的相互作用密切相关[18-21]。即AD退行性进展与其信号通路的改变有关。细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)信号通路是经典MAPK信号通路成员之一,其成员包括ERK1和ERK2,分子量分别为44 kD和42 kD[22]。有研究发现,ERK在中枢神经系统中对神经细胞增殖和分化、突触可塑性、学习记忆能力和轴突生长等具有重要意义[23]。据报道,ERK主要存在神经元细胞的轴突、树突中,其通路激活(即ERK磷酸化)可以将刺激信号传导到细胞核内,且ERK信号通路参与了AD的病理发展过程,在APP的加工过程中发挥重要作用[24]。Pak等[20]和Hong等[21]最新研究发现,ERK(正常调控范围下)能激活细胞外信号调节激酶(cAMP)反应元件结合蛋白信号通路(ERK/CREB),进而激活Ras-MAPK通路促进神经元生长再生。另外,Sun等[22]的研究表明,ERK信号通路激活能增加α-分泌酶的活性,进而促进可溶性APPα释放,减少Aβ的产生和沉积。
为探讨Aβ1-42导致HT22分化及凋亡的机制,我们通过分析ERK与Aβ及凋亡基因的相关性来分析可能的靶向信号通路,结果显示:在基因组水平上,ERK与APP,CASP9表达呈正相关。而Caspase-9为Caspase-3的激动剂,可以通过促进Caspase-3剪切成cleaved-Caspase-3激活细胞凋亡机制。据报道,分子层面上,Aβ斑块可刺激促进神经元中与细胞周期进展相关的因子的异常表达,从而导致Caspase-3激活增加,促进神经元凋亡[25]。为进一步明确Aβ、ERK与凋亡的相关性,我们通过Western blot发现AD组ERK磷酸化比例增高,即ERK信号通路处于高激活状态。现有研究报道,AD细胞中多个信号通路共同作用,具体表现为:Ras-ERK信号通路参与细胞周期和凋亡的进展、JNK信号通路与抗凋亡蛋白表达呈负相关、ERK信号通路磷酸化与不可溶性蛋白沉积密切相关、JAK-STAT信号通路增强Caspase-3介导的神经变性[4,26]。因此,我们推测Aβ1-42可能增强Ras-ERK信号级联,而ERK信号通路很可能为Aβ1-42导致HT22细胞凋亡、阻滞细胞分裂,分化为AD细胞的靶向通路之一。
结合众多研究,我们认为ERK通路对AD进展可能具有轻度抑制作用。但由于AD病程不可逆性的加剧,APP基因调控生成Aβ斑块的能力可能大于ERK通路清除Aβ斑块的能力,故ERK在一定程度上可能延缓AD进展,但并不能逆转AD进程。因此我们推测:AD病程不可逆性的加剧可能与ERK通路磷酸化程度有关,即p-ERK/ERK比值。然而,ERK是否在AD进程中发挥抑制作用,以及ERK在AD进程中发挥作用的具体机制仍未完全阐明。因此,未来我们将把更多精力投入在AD细胞模型中ERK信号通路相关蛋白的监测上,以期通过荧光显微镜技术等标记信号分子和细胞结构,通过活细胞成像实时观察信号分子的动态变化。
综上所述,在Aβ1-42浓度为20 μmol/L,干预时间为24 h的培养条件下,HT22细胞表现出较明显的AD病理特征,可以认为是较理想的AD细胞建模条件。同时,本实验利用生物信息学的技术,分析并验证Aβ1-42导致神经损伤及细胞凋亡的候选靶向信号通路之一是ERK,继而为探索AD的发病机制提供新的思路,对于在细胞水平和分子水平上研究AD的发病机制提供科学依据。
猜你喜欢
自然杂志(2021年6期)2021-12-23
建材发展导向(2021年11期)2021-07-28
当代水产(2020年10期)2020-03-17
当代水产(2019年8期)2019-10-12
现代装饰(2018年5期)2018-05-26
中国病理生理杂志(2015年8期)2015-12-21
电源技术(2015年5期)2015-08-22
医学研究杂志(2015年3期)2015-06-10
弹箭与制导学报(2015年1期)2015-03-11
创业家(2015年1期)2015-02-27
