
婴儿β-酮硫解酶缺乏症1例
2024-03-22 12:02蔺艳婷李坤霞陈坤琦张爱梅山东第二医科大学临床医学院儿科教研室潍坊605烟台毓璜顶医院儿内科潍坊市人民医院新生儿科通讯作者mailzamcncom
山西医科大学学报 2024年2期
蔺艳婷,李坤霞,陈坤琦,张爱梅(山东第二医科大学临床医学院儿科教研室,潍坊 605;烟台毓璜顶医院儿内科;潍坊市人民医院新生儿科;通讯作者,E-mail:zamcn@6.com)
β-酮硫解酶缺乏症(β-ketothiolase deficiency,BKD),又称线粒体乙酰乙酰基辅酶A硫解酶(T2)缺乏症,是一种罕见的由乙酰辅酶A酰基转移酶-1(acetyl coenzyme A acetyltransferase-1,ACAT1)基因变异引起的常染色体隐性遗传代谢性疾病。ACAT1基因编码在人类染色体11q22.3-23.1上,横跨基因组约27 kb的区域,包含12个外显子和11个内含子[1]。T2缺乏的临床特征是不同程度的间歇性酮症酸中毒发作,严重者可能出现认知功能损害、神经系统发育落后甚至死亡。现回顾性分析1例经基因诊断确诊的BKD婴儿临床资料,探索BKD患儿的临床表现及遗传特点,进一步提高临床医师对该病的认识。
1 病例报告
1.1 病例资料
患儿,女,7月,以“呼吸困难1 d”入院。患儿1 d前无明显诱因出现呼吸困难,表现为呼吸幅度增大,伴呻吟及轻咳,吃奶少,无发热,无呕吐、腹胀、腹泻,无抽搐及意识障碍。患儿系G2P2,父母体健,无遗传病病史。有1个哥哥,6岁,体健。
入院体格检查:呼吸32次/min,心率142次/min,SpO298%,体质量7.5 kg。发育正常,营养欠佳,嗜睡,轻度脱水貌。深大呼吸,双肺呼吸音粗,未闻及干湿性啰音。心音有力,律齐,各瓣膜听诊区未闻及杂音。腹平软,肝脏肋下2 cm,脾脏肋下1 cm,肠鸣音正常。
入院辅助检查:①血气分析:pH 6.951,PCO210.6 mmHg,PO2130 mmHg,ABE -30.0 mmol/L,SBE -27.7 mmol/L,实际碳酸氢根2.2 mmol/L,标准碳酸氢根5.3 mmol/L,乳酸1.6 mmol/L;②血常规:红细胞4.21×1012/L,白细胞32.54×109/L,中性粒细胞百分比73.9%,血小板462×109/L;③尿常规:尿蛋白+,尿酮体+++;④总胆固醇5.83 mmol/L(参考值3.12~5.72 mmol/L),甘油三酯2.01 mmol/L(参考值0.40~1.70 mmol/L);⑤甲功:FT4为6.79 pmol/L(参考值12.00~22.00 pmol/L),FT3为1.56 pmol/L(参考值2.8~7.1 pmol/L);⑥胸片:未见异常;⑦颅脑MR:右侧基底节区血管间隙扩大。
串联质谱检测结果:血串联质谱遗传代谢病检测提示患儿3-羟基异戊酰肉碱(C5-OH)为1.18 μmol/L(参考值0.05~0.70 μmol/L)、异戊烯酰肉碱(C5∶1)为0.24 μmol/L(参考值0.01~0.06 μmol/L)、3-羟基丁酰肉碱(C4-OH)为1.66 μmol/L(参考值0.03~0.75 μmol/L),均显著增高。尿气相色谱质谱有机酸检测提示患儿2-甲基-3-羟基丁酸为22.3 μmol/L(参考值0~4.0 μmol/L)、3-甲基巴豆酰甘氨酸为24.7 μmol/L(参考值0~5.0 μmol/L)、甲基巴豆酰甘氨酸为15.3 μmol/L(参考值0~5.0 μmol/L),均明显增高。
1.2 基因检测
本研究经医院医学伦理委员会批准(KYLL20230904-4)并获得患儿监护人知情同意并签署知情同意书。采集患儿外周血样3 mL,提取全血基因组DNA,制备文库,靶向捕获与富集,利用NovaSeq 6000测序仪进行高通量测序。对候选变异在HGMD、1000 Genomes、dbSNP、gnomAD、ClinVar、ESP 6500等数据库检索收录情况。依据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异评级指南对候选变异致病性进行判读。结果提示患儿ACAT1(NM_000019.3)基因4号外显子存在c.253_255del(p.Glu85del)杂合变异,8号外显子存在c.806_807del(p.Thr269fs)杂合变异(见图1),均为疑似致病变异。
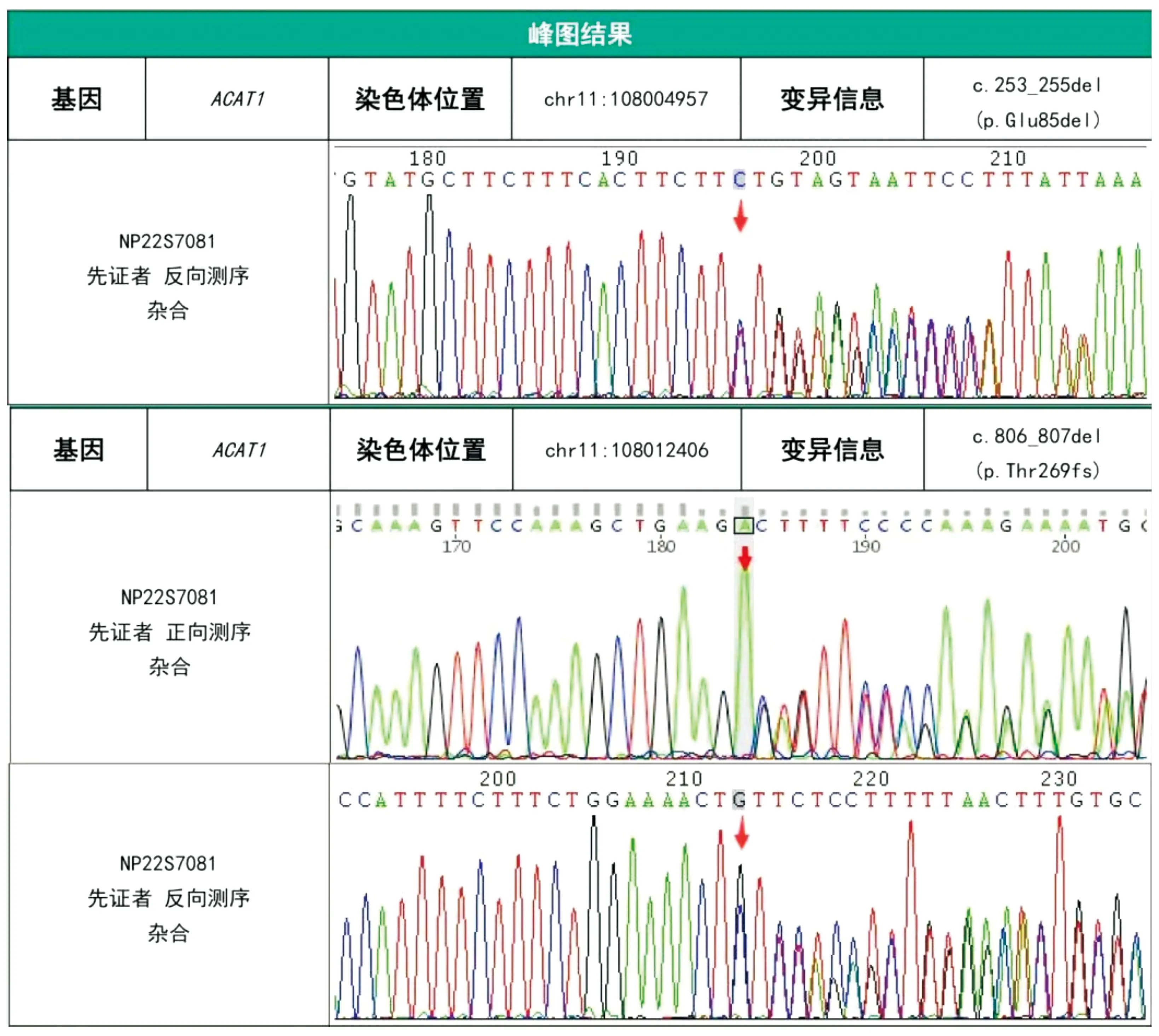
图1 患儿基因检测结果
1.3 治疗与随访结果
该婴儿入院后给予头孢哌酮舒巴坦钠抗感染,碳酸氢钠纠酸及补液治疗。住院期间出现抽搐1次,给予水合氯醛灌肠止痉,急查颅脑CT未见异常。遗传代谢病检查结果提示β-酮硫解酶缺乏,给予禁蛋白饮食,无亮氨酸营养粉喂养,加左卡尼汀促进酸性代谢产物排出。患儿住院治疗9 d,呼吸困难缓解,复查血气分析大致正常。出院诊断为:β-酮硫解酶缺乏症;重度代谢性酸中毒;代谢性脑病;高甘油三酯血症;急性支气管炎。出院后予以适当限制蛋白质摄入量,高热量、低脂饮食,无亮氨酸营养粉喂养,口服左卡尼汀治疗,随访患儿至2岁无酸中毒发作,生长发育正常。
2 讨论
β-酮硫解酶缺乏症是一种罕见的异亮氨酸分解代谢和酮体代谢异常的先天性疾病。其发病年龄通常为6~18个月。典型表现为由禁食、感染或体力消耗等生酮应激状态下引起的呕吐、呼吸困难、呼吸急促、嗜睡或昏迷[2]。在发病前及时干预和治疗,可完全恢复或延缓发病时间。BKD的实验室特征性表现包括显著的酮尿和尿中异亮氨酸分解代谢中间产物2-甲基-3-羟基丁酸、2-甲基乙酰乙酸和甲基巴豆酰甘氨酸的排泄增加。血串联质谱检测通常显示异戊烯酰肉碱和3-羟基异戊酰肉碱水平升高。有研究表明3-羟基丁酰肉碱同样是检测BKD的一个非常强大的标记物[3]。
本例患儿因呼吸困难入院,血气分析示重度代谢性酸中毒合并失代偿性呼吸性碱中毒,尿常规示酮体+++,住院期间出现抽搐,血串联质谱示C5-OH、C5∶1、C4-OH显著增高,尿气相色谱质谱示2-甲基-3-羟基丁酸、3-甲基巴豆酰甘氨酸、甲基巴豆酰甘氨酸明显增高,确诊为β-酮硫解酶缺乏症。基因检测示患儿ACAT1基因4号外显子c.253_255del为框内缺失变异,导致所编码蛋白质第85位氨基酸Glu发生缺失,但不会引起移码。有文献报道在乙酰乙酰基辅酶A硫解酶缺乏症患者中检测到该变异[1],该变异在大规模人群频率数据库gnomAD中有2例杂合子报道,未见纯合子报道。功能学研究表明,该变异导致线粒体乙酰乙酰基辅酶A硫解酶活性显著降低。依据ACMG指南评级为疑似致病变异(PM2+PM4+PS3_Moderate+PM3_Supporting)。ACAT1基因8号外显子c.806_807del是非3倍数碱基缺失导致的移码变异,理论上可通过无义介导的mRNA降解(NMD)或编码氨基酸序列的提前终止,而导致正常蛋白功能丧失。该变异在HGMD、dbSNP、ESP 6500、1000 Genomes等数据库均未见收录。依据ACMG指南评级为疑似致病变异(PVS1+PM2)。患儿父母拒绝对基因变异进行溯源,因此不能明确突变来源。患儿父母临床表型均未见异常,考虑为隐性携带者的可能性大。我们推测患儿的框内缺失变异及移码变异分别来自父母,结合患儿临床表型、家族史、串联质谱检测及基因检测结果,考虑患儿为ACAT1基因复合杂合突变所致的β-酮硫解酶缺乏症。
BKD的发病率各个国家报道不一,无明显的种族特异性,越南北部的发病率估计为1∶190 000[4],北卡罗来纳州为1∶313 000[3],美国明尼苏达州为1∶232 000[5]。中国的报道多为散发病例,少见流行病学的相关分析资料。在浙江省新生儿疾病筛查中心的一项多中心国家队列研究估计中国的发病率最低约为1∶1 000 000。迄今为止,至少有105种与BKD相关的ACAT1基因变异报道[6]。最常见的变异类型是错义突变。基因变异的类型在不同国家和种族之间有所不同。越南北部的热点变异为c.622C>T(p.Arg208X)[7],印度的热点变异为c.578T>G(p.Met193Arg)[4]。徐烽等[8]报道c.1124A>G(p.Asn375Ser)变异可能是中国人的热点变异。值得一提的是,本例患儿c.806_807del变异在中国知网及PubMed等数据库均未见相关报道,为新发变异。
ACAT1基因编码的线粒体乙酰乙酰辅酶A硫解酶(T2)在异亮氨酸分解中使2-甲基-乙酰乙酰辅酶A分解为乙酰辅酶A和丙酰辅酶A,T2在酮体利用过程中催化乙酰乙酰辅酶A生成2分子乙酰辅酶A[9]。BKD患儿T2活性降低或丧失,异亮氨酸分解代谢障碍,导致大量酸性代谢产物蓄积,同时大量酮体在组织细胞中聚集。因此患儿常反复发生严重代谢性酸中毒及多脏器损害。本例患儿血气分析示重度代谢性酸中毒,因诊断及治疗及时,尚未发现有严重的脏器损害。因此,早筛查、早诊断及合理治疗是挽救患儿、改善预后的关键。我们要积极普及新生儿遗传代谢性疾病筛查,以期在急性代谢性酸中毒首次发作之前得到及时、恰当的诊治。我们应该警惕出现严重代谢性酸中毒、不明原因的昏迷或神经系统发育落后的患儿发生遗传代谢性疾病的可能,及时送血尿标本行串联质谱检测,以尽早明确诊断。
目前对于无症状或非发作性症状的BKD患儿,应避免禁食,建议口服L-肉碱,补充剂量为50~200 mg/(kg·d)。若患儿处于生酮状态,如呕吐、食欲减退或感染等,即使血糖正常,也应口服或静滴补充充足的葡萄糖,以抑制酮体的生成。在密切监测血糖的同时,使用胰岛素(1 U)与足量的葡萄糖(4~6 g)配制静脉输注,可预防患儿发生严重的酮症酸中毒[10]。同时根据个体情况补充碳酸氢钠。严重患儿需要血液透析或血浆置换。处于稳定期的患儿需限制蛋白质。有文献报道[11],建议婴儿蛋白质摄入量控制在1.5~2.0 g/(kg·d),1岁以上的患儿蛋白质摄入量控制在1.0~1.5 g/(kg·d)。同时注意避免高脂饮食[12]。口服左卡尼汀有助于稳定代谢状况,左卡尼汀可与有机酸代谢产物形成酰基肉碱,有利于有机酸的排泄。
关于基因型与表型的关系,以往的研究表明,基因型与临床表型在BKD中并不相关。即使BKD患者具有相同的基因型和相似的环境因素,也可能具有不同的临床表型。虽然基因型可能影响BKD患者的生化表型,但由于缺乏对大量家族性ACAT1变异的研究,基因型与生化表型之间的关系难以分析[3]。
综上所述,本研究通过基因检测对1例BKD婴儿进行了精准的分子学诊断,确诊其为ACAT1基因c.253_255del及c.806_807del复合杂合变异所致的β-酮硫解酶缺乏,为该婴儿的后续治疗及家系遗传咨询提供了理论依据。新变异的检出同时拓展了ACAT1基因的变异谱。
猜你喜欢
中老年保健(2022年2期)2022-11-25
中老年保健(2022年4期)2022-08-22
生命科学研究(2022年2期)2022-05-15
昆明医科大学学报(2022年1期)2022-02-28
中国畜牧杂志(2019年4期)2019-04-20
家庭百事通·健康一点通(2017年3期)2017-03-22
老年医学与保健(2017年6期)2017-02-06
中外医疗(2016年15期)2016-12-01
动物营养学报(2015年9期)2016-01-07
中外医疗(2015年11期)2016-01-04
