
极光激酶A通过促进巨噬细胞源性泡沫细胞形成参与动脉粥样硬化发生发展
2024-03-25 04:33耿戈戈张峰华刘晓炎樊雯婷
赣南医学院学报 2024年1期
耿戈戈,张峰华,刘晓炎,樊雯婷,甘 滔
(1. 赣南医科大学基础医学院;2. 赣南医科大学康复学院,江西 赣州 341000)
动脉粥样硬化(Atherosclerosis,AS)是一种常见的慢性病,涉及多种严重并发症,是冠心病、脑梗死、外周血管病发病的主要因素。无论是在发达国家还是发展中国家,AS的发病率与死亡率都在逐年升高,严重危害人类健康。AS的发病受多种因素影响,包括高胆固醇血症、高血压、糖尿病和吸烟等[1]。AS的发病过程主要涉及血管内皮细胞、平滑肌细胞和巨噬细胞的一系列病变,多项研究表明,这些细胞参与了动脉粥样硬化斑块的形成,抑制其病变的发生是改善AS 的重要着手点[2-4]。在AS 发生过程中,血管内皮细胞可被氧化后的脂蛋白或其他炎症分子激活,通过表达血小板选择素(P-selection)、内皮细胞选择素(E-selection)等分子进一步招募单核细胞[5]。被招募的单核细胞在进入血管内膜区域后受巨噬细胞集落刺激因子(Macrophage colony-stimulating factor, M-CSF)等细胞因子刺激分化为巨噬细胞,后者通过清道夫受体吸收氧化低密度脂蛋白进一步转变成泡沫细胞。泡沫细胞在经历频繁的凋亡或坏死后逐步累积形成坏死核,其中包含胆固醇脂、胆固醇结晶以及细胞碎片,显著增加了斑块破裂的可能[6]。随着斑块的形成,血管中层的平滑肌细胞进入增殖状态并迁移至内膜,随后分泌主要由胶原构成的细胞外基质并形成保护性纤维帽。位于纤维帽中的平滑肌细胞能够演变成巨细胞样,通过吸收氧化脂蛋白发生泡沫化并随之凋亡,加剧了斑块内的坏死和炎症水平,增加了斑块的不稳定性[7]。目前临床上针对AS 的治疗方法主要是降脂治疗,例如他汀类药物、前蛋白转化酶枯草溶菌素/Kexin9 型(PCSK9)抑制剂和非他汀类降脂药物,以及一些抗炎药物和血小板药物的抑制性治疗,但以上药物都无法达到理想的治疗效果,甚至还伴有较严重的不良反应[8-9],因此寻找新的有效靶点势在必行。
极光激酶A(Aurora kinase A, AurkA)是一种丝氨酸/苏氨酸激酶,在细胞的有丝分裂中起着重要作用,是细胞周期调控的关键基因。除了在有丝分裂中起作用,AurkA 过量表达时还可作为癌基因促进癌症的发生和发展。研究发现,AurkA 通过调控增殖、上皮-间质转化、转移、凋亡以及癌症干细胞的自我更新等促进癌症发生[10-12]。现有多种针对AurkA的特异性小分子抑制剂被开发并且进入临床试验阶段,使AurkA 成为高度可行的治疗靶点[13]。除癌症之外,AurkA 参与糖尿病、主动脉夹层动脉瘤、肺动脉高压等多种代谢相关疾病的调控[14-16]。在糖尿病模型小鼠中,抑制AurkA 可有效减少巨噬细胞在胰岛组织中的浸润并且显著下调巨噬细胞中促炎分子IL-6 的分泌[17],提示AurkA 可能调节促炎性巨噬细胞的生成。DING L 等[18]研究发现,AurkA 高表达于M1 型巨噬细胞,其酶活的抑制能有效遏制M1极化,阻碍自身免疫性脑炎等炎性疾病的发生。AurkA 作为抗坏死因子能够特异性地促进M1 型巨噬细胞存活,破坏炎性/抗炎性巨噬细胞平衡[17]。同时,关于肺动脉高压以及主动脉夹层动脉瘤等血管疾病的相关研究显示,AurkA 能够调控平滑肌细胞功能,包括增殖和迁移[15]。由此可见,AurkA 与巨噬细胞以及平滑肌细胞的功能调控密切相关。基于此,我们猜测AurkA可能通过调控巨噬细胞、平滑肌细胞等多种细胞参与AS发生发展。因此,本研究通过动物模型和细胞实验初步探究AurkA在动脉粥样硬化发生发展中的潜在作用及可能机制。
1 材料与方法
1.1 实验材料 C57BL/6J 小鼠(北京维通利华公司),IMDM 培养基(Gibco 公司),氧化低密度脂蛋白(广州奕元生物技术有限公司),油红染色液(Solarbio 公司),AurkA 抑制剂MLN8237 以及TCS7010(Selleck 公司),2-羟丙基-β-环糊精(Selleck公司),甘油三酯测试盒(南京建成生物工程研究所),Murine M-CSF(Peprotech 公司),Murine IFN-γ(Peprotech公司),Murine LPS(Sigma-Aldrich公司)。
1.2 实验方法
1.2.1 动脉粥样硬化小鼠病灶区基因表达谱分析 从GEO 数据库提取动脉粥样硬化发病小鼠主动脉基因表达谱数据(GSE 19286),包括了动脉粥样硬化发病小鼠主动脉样本(Aorta_APOE-deficient with atherosclerosis_1 和Aorta_APOE-deficient with atherosclerosis_2)以及未发病对照小鼠主动脉样本(Aorta_non-transgenic C57BL/6J control_1 和Aorta_non-transgenic C57BL/6J control_2)的基因表达数据,选择特异性识别AurkA 的探针1424511_at和1440129_at,分析AurkA 表达。AurkA 在各类血液细胞中的表达谱数据来自Bloodspot 数据库(https://fobinf.com),选择的数据集为Mouse Normal (RNA-Seq)。
1.2.2 动物模型构建 ⑴AurkA 表达验证动物模型构建:选用8 周龄普通饲料喂养的C57BL/6J 健康雄性小鼠作为CC组(对照组),再选用8周龄C57BL/6J背景的ApoE-/-(Apolipoprotein E KO)健康雄性小鼠,随机分为AN 组及AH 组。CC 组和AN 组喂养普通饲料,AH 组喂养21%脂肪、0.5%胆固醇的高脂饲料,每2 天更换1 次饲料,每周进行体重监控,共喂养16 周构建动脉粥样硬化小鼠模型;⑵AurkA 抑制剂功能实验动物模型构建:选用8 周龄C57BL/6J 背景的ApoE-/-健康雄性小鼠,随机分为MC 组(MC 为溶剂处理的高脂喂养的ApoE-/-小鼠组)和M 组(AurkA 抑制剂MLN8237 处理的高脂喂养的ApoE-/-小鼠组),以同样方法高脂喂食16 周构建动脉粥样硬化小鼠模型,在建模第12 周开始M 组给予小鼠20 mg·kg-1MLN8237 灌胃,每周2 次,持续4 周,MC组给予小鼠同等体积溶剂(10% 2-羟丙基-β-环糊精和1%碳酸氢钠)处理。
1.2.3 血脂测定 将全血室温静置30~60 min,有明显的分层后,取血清直接测定,如超过线性范围用生理盐水稀释后测定。按照血清总胆固醇(Total cholesterol,TC)检测试剂盒、甘油三酯(Triglycerides,TG)检测试剂盒以及低密度脂蛋白胆固醇(Low density lipoprotein cholesterol,LDL-C)检测试剂盒说明书指示,分别设置空白孔、标准孔、样本孔。空白孔加入2.5 μL蒸馏水,标准孔加入2.5 μL 校准品,样本孔加入2.5 μL样本液,每个孔中都加入250 μL的工作液,随后振荡孔板混匀,37 ℃孵育10 min,测定波长500 nm 处各孔吸光度。
1.2.4 油红染色 ⑴小鼠主动脉大体油红染色:称取0.5 g 油红加入100 mL 异丙醇中,过夜溶解后使用滤纸过滤直至无沉淀,将配制成的油红原液按油红∶超纯水=3∶2 混合,配制成油红工作液备用。将剥离完的主动脉放入4%多聚甲醛溶液中避光固定72 h,将固定好的主动脉放入超纯水中漂洗10 min,再转移至60%异丙醇中漂洗10 min 后转至油红工作液中染色60 min。将小鼠主动脉转移到60%异丙醇中漂洗1 min,弃去旧液,加入新的异丙醇重复洗3次,最后将小鼠主动脉转移到超纯水中,染色完成;⑵主动脉根部油红染色:将固定完毕的小鼠心脏组织依次放于15%、40%的蔗糖溶液内进行脱水,将脱好水的组织取出吸干表面水分,用镊子轻轻将组织放入包埋台进行包埋,待OCT(Optimal cutting temperature)将组织完全覆盖后将包埋台水平放于干冰上,快速冷冻直至OCT 变白变硬后再进行切片。将主动脉根部冰冻切片复温并干燥10 min,4%多聚甲醛固定15 min 后清洗晾干,随后加入油红工作液避光染色20 min,染色结束后使用60%异丙醇对切片浸洗2 次,每次5 min,脱色至背景清晰,蒸馏水浸洗3次;苏木素染色5 min,水洗3次后晾干并用甘油封片即可;⑶泡沫细胞油红染色:收取BMDM接种于96 孔板培养,加入氧化低密度脂蛋白(Oxidized low-density lipoprotein,ox-LDL)诱导泡沫化,培养24 h后吸掉原培养液,PBS洗2次,加入油红固定液,避光常温30 min,然后弃液用ddH2O洗2次,加入60%异丙醇洗5 min,弃去异丙醇,加入油红染液,摇晃避光染色30 min,弃去油红染液,水洗2~5次,加入ORO buffer,浸泡1 min,弃液,加入ddH2O,覆盖细胞,显微镜下观察并拍照。
1.2.5 实时荧光定量聚合酶链式反应(Real-time quantitative PCR,qPCR) 使用Trizol法提取总RNA,检测RNA 浓度和纯度后进行逆转录,所得cDNA 用于进行实时定量PCR实验。吸取10 μL TB GreenⅡ、1 μL 上、下游引物、1 μL cDNA,用RNase free water补足到20 μL上机进行PCR反应,反应条件为:95 ℃预变性20 min,95 ℃变性10 min,61 ℃退火20 s,72 ℃延伸25 s,共40个循环。以β-actin 作为内参基因,AurkA 引物序列:上游引物5′-CTGGATGCTGCAAACGGATAG-3′,下游引物5′-CGAAGGGAACAGTGGTCTTAACA-3′。β-actin 引物序列:上游引物5′-AACAGTCCGCCTAGAAGCAC-3′,下游引物5′-CGTTGACATCCGTAAAGACC-3′。
1.2.6 HE 染色 先将主动脉根部切片,4%多聚甲醛固定15 min,苏木素染色5 min,自来水冲洗干净,85%、95%的酒精脱水5 min,伊红染色5 min,依次放入无水乙醇Ⅰ 5 min→无水乙醇Ⅱ 3 min→无水乙醇Ⅲ 3 min→二甲苯Ⅰ3 min→二甲苯Ⅱ 3 min直至脱水透明,晾干,封片。
1.2.7 骨髓源性巨噬细胞(Bone marrow-derived macrophage,BMDM)培养 使用8 周龄C57BL/6J 健康雄性小鼠,断颈处死,从股骨和胫骨中冲洗出骨髓细胞,经红细胞裂解过滤后制备细胞悬液,加入12 μL M-CSF(终浓度为10 ng·mL-1)进行铺板培养,72 h 后补加6 mL 培养液与6 μL M-CSF(终浓度为 10 ng·mL-1),再培养4 d刺激BMDM。
1.2.8 巨噬细胞泡沫化 BMDM 诱导成功后,收集细胞重新铺板培养,分为对照组(Con)、建模组(ox-LDL)以及AurkA 抑制剂处理组(ox-LDL+TCS)。在2 组细胞中加入100 μg·mL-1ox-LDL 构建泡沫细胞模型,同时给予ox-LDL+TCS 组10 nM AurkA 抑制剂TCS7010处理,给予ox-LDL组等量溶剂处理,48 h后利用油红染色检测泡沫细胞的脂质累积情况。
1.2.9 流式细胞分析仪检测 收集外周血细胞,用含2% FBS 的PBS 制成单细胞悬液,300 g 离心5 min,弃掉上清液,加入红细胞裂解液,用10 mL 细胞染色Buffer 终止红细胞裂解,300 g 离心5 min,弃掉上清,将细胞制成107·mL-1悬液,取100 μL加入相应荧光标记的抗体(100∶1 或200∶1),混匀后置于4 ℃,避光孵育20 min,洗涤后重悬细胞,用流式细胞仪进行检测,并利用Flowjo软件进行数据分析。
1.3 统计学处理 采用Prism 8.0 统计软件对数据进行分析,各组数据均用均数±标准差表示,组间两两比较采用t检验,组间多重比较采用单因素方差分析(One-way ANOVA)检验。检验水准α=0.05。
2 结果
2.1 动脉粥样硬化斑块病灶区AurkA 的表达 从GEO 数据库中调取AS 造模小鼠病灶区基因芯片数据分析,与CC 组相比,AH 组动脉粥样硬化斑块病灶区AurkA 表达升高(图1),提示AurkA 高表达与AS发病相关。
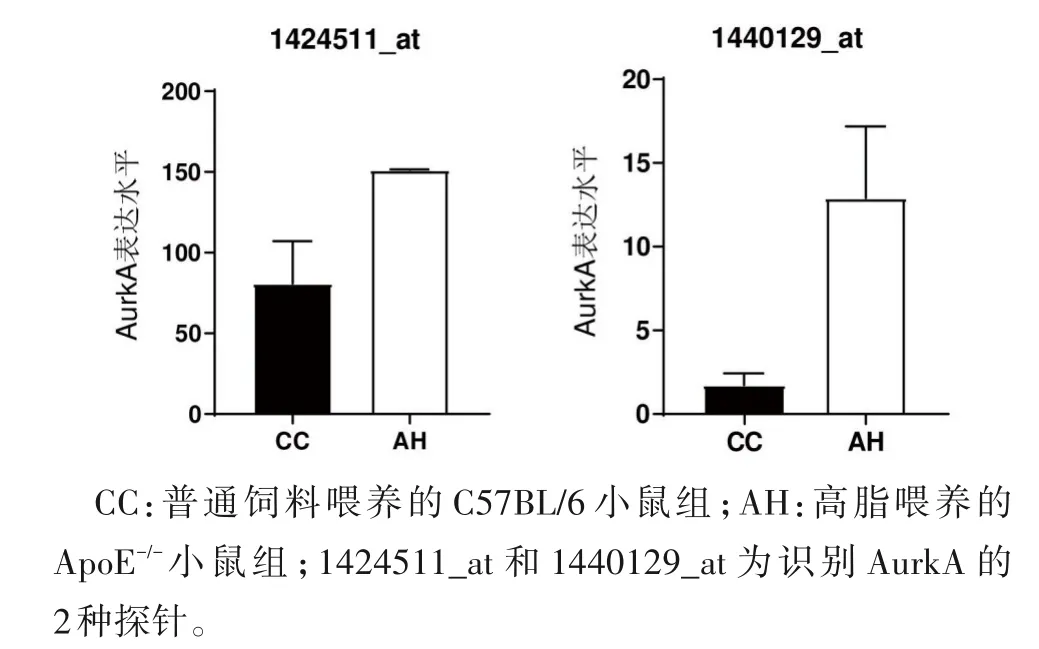
图1 动脉粥样硬化斑块病灶区AurkA的表达
2.2 主动脉中的粥样斑块分布情况及血清TC、TG和LDL-C含量 主动脉大体油红染色结果显示AH 组主动脉中的粥样斑块分布明显多于AN 对照组(P<0.05)(图2);AH 组血清中TC、TG 和LDL-C高于AN 组和CC 组,AN 组血清中TC、LDL-C 高于CC 组,差异均有统计学意义(P<0.05)(图3)。提示AS模型构建成功。

图2 主动脉中的粥样斑块分布情况

图3 小鼠血清TC、TG和LDL-C含量比较
2.3 小鼠肝脏、脾脏和主动脉组织AurkA mRNA表达 与AN组相比,AH组肝脏、脾脏和主动脉组织AurkA mRNA 增加,差异均有统计学意义(P<0.05)(图4)。
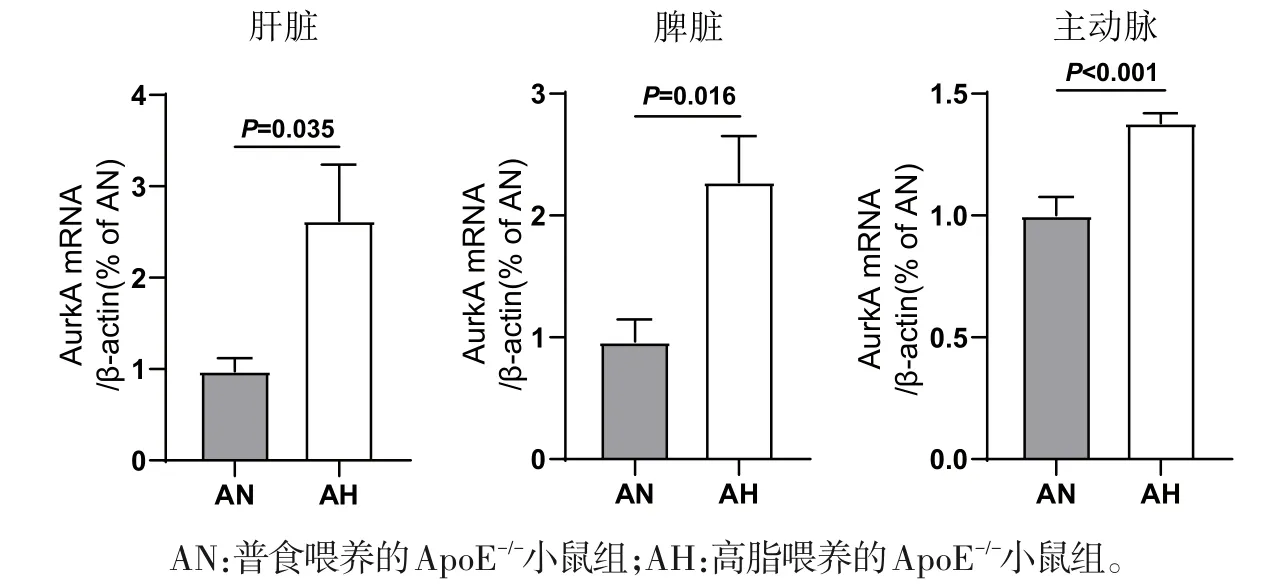
图4 小鼠肝脏、脾脏和主动脉组织AurkA mRNA表达
2.4 AurkA 抑制剂对小鼠体重以及血清TC、TG、LDL-C 含量的影响 与MC 组相比,M 组小鼠体重及血清TC、TG、LDL-C 水平无明显差异(P>0.05)(图5)。

图5 AurkA抑制剂对小鼠体重以及血清TC、TG、LDL-C的影响
2.5 小鼠主动脉斑块比较 2 组小鼠主动脉内壁斑块总面积无明显差异(图6),与MC 组比较,M 组主动脉窦中斑块面积及油红面积减小,差异均有统计学意义(P<0.05)(图7)。
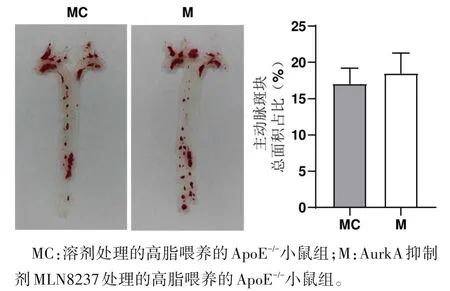
图6 AurkA抑制剂对小鼠主动脉斑块总面积的影响

图7 AurkA抑制剂对小鼠主动脉根部斑块面积的影响
2.6 AurkA 调控巨噬细胞源性泡沫细胞形成 流式细胞术分析结果显示,AurkA抑制剂处理虽未改变外周血中髓性细胞占比,但能显著下调外周血中泡沫细胞前体,即单核细胞的占比(P<0.05)(图8)。通过对各类血细胞中AurkA mRNA 分析发现,髓性细胞尤其是巨噬细胞中存在AurkA mRNA 的特异性高表达(图9),BMDM 的泡沫细胞体外诱导实验发现,相较于未处理的对照组,BMDM 经100 μg·mL-1的ox-LDL刺激后,大量累积脂质形成泡沫细胞(图9)。加入AurkA酶活抑制剂处理后,细胞中脂滴累积程度相较造模组显著降低(P<0.05)(图10)。
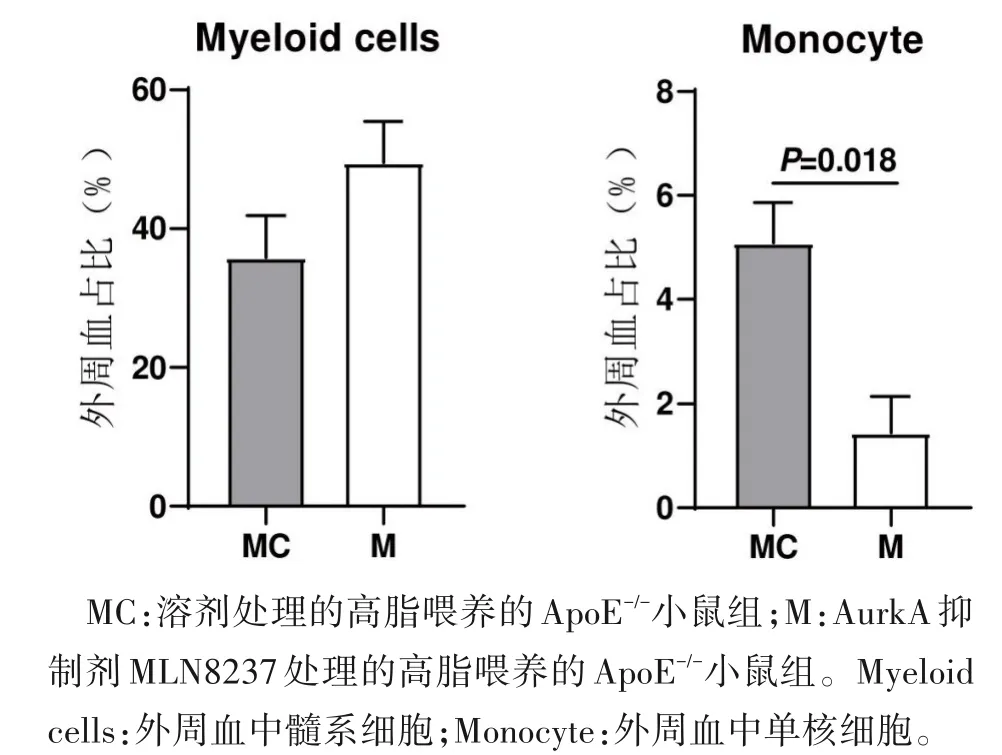
图8 AurkA 抑制剂对动脉粥样硬化小鼠外周血中髓系细胞占比的影响

图9 不同血细胞AurkA mRNA的表达
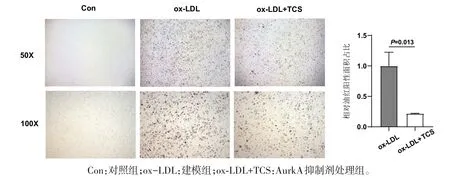
图10 AurkA对巨噬细胞源性泡沫细胞生成的影响
3 讨论
动脉粥样硬化是一种常见的慢性疾病,也是全球人口死亡的主要原因,其发病机制复杂,涉及多种细胞相互作用,近年来不断有研究表明可以通过调控单种或多种细胞促进或抑制AS的发生发展,但适用于临床转化且易于靶向的靶点研究较少。
AurkA 作为调控细胞周期的关键分子,其表达或功能异常与疾病的发生息息相关。前期研究大多集中于AurkA 在癌症发生发展中的作用,而对其是否参与非癌症疾病的发生研究较少。近年来,AurkA 被发现与多种慢性炎症以及代谢疾病的发生相关。在实验性自身免疫性脑炎动物模型中,AurkA通过促进巨噬细胞的M1 极化提高炎症水平促进疾病发展[18];在胰岛素模型小鼠中,抑制AurkA能够显著减少巨噬细胞在病灶区的浸润、下调促炎因子IL-6水平并改善胰岛素抵抗,从而缓解发病[19];同样通过减少促炎因子表达水平,AurkA 抑制剂显著减轻了滑膜炎症从而缓解化脓性关节炎[19]。本研究首先通过基因芯片数据发现了AurkA 高表达于AS 斑块病灶区,随后通过动物模型验证AurkA与AS发病的相关性。利用小分子抑制剂抑制AurkA 后发现,AS模型小鼠血脂水平不受影响,主动脉斑块总面积无变化,但主动脉窦斑块厚度显著减少,斑块中脂质累积明显降低,同时AurkA被抑制后,外周血中的单核细胞数量减少,进而限制了斑块中单核细胞的浸润以及向巨噬细胞的转化。斑块中巨噬细胞的减少抑制了泡沫细胞的形成,同时细胞实验发现抑制AurkA能够减少巨噬细胞中的脂质累积从而限制巨噬细胞源性泡沫细胞的生成。因此,AurkA 通过抑制巨噬细胞源性泡沫细胞的生成,进而控制斑块形成以及脂质堆积,改善动脉粥样硬化。这一发现与前期研究相一致,提示了AurkA 与巨噬细胞功能调控紧密相关。此前AurkA 被发现通过激活AKT信号通路实现对PPARα 以及PGC1α 等脂质代谢基因的调控[20-21];同时在巨噬细胞中抑制AurkA 活性可影响IκBα 磷酸化水平,抑制NF-κB 信号通路激活[18],而后者对于泡沫细胞形成有显著的促进作用[22];另有研究[23]发现,在肺癌细胞中AurkA 可调控CXCL5的基因水平,而CXCL5同样可以限制动脉粥样硬化中巨噬细胞泡沫细胞的形成。因此,AurkA可能通过多通路多靶点调控巨噬细胞功能,影响其脂质代谢水平和炎症水平,参与泡沫细胞形成。同时,我们还发现AurkA能够影响外周血中的单核细胞水平。有研究[24]表明,在AS患者以及造模小鼠中都能观察到髓系造血水平的增强,其中外周血中单核细胞数量增加对斑块生成具有显著促进作用。由此可见,抑制AurkA 功能不仅直接影响巨噬细胞向泡沫细胞转化,同时还从源头上减少侵入血管内膜的巨噬细胞数量,两种机制共同作用抑制斑块形成。
综上所述,本研究通过动物模型和细胞实验证明了AurkA 高表达与AS 相关,并且发现AurkA 通过调控巨噬细胞源性泡沫细胞生成参与AS发生发展。AurkA 作为蛋白激酶,其酶活可以被特异性靶向。目前已有多种针对AurkA的小分子抑制剂被开发并进入临床试验阶段,包括本研究中所使用的MLN8237。因此,本研究为动脉粥样硬化的临床治疗提供了一个高度可行的新靶标,也为相关药物开发提供了新的思路和理论基础。
猜你喜欢
环球时报(2023-03-22)2023-03-22
作文周刊·小学一年级版(2022年20期)2022-05-07
昆明医科大学学报(2022年2期)2022-03-29
趣味(数学)(2021年4期)2021-08-05
临床与实验病理学杂志(2020年9期)2020-11-03
天津医药(2016年9期)2016-10-20
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国比较医学杂志(2015年5期)2016-01-28
创业家(2015年9期)2015-02-27
哈尔滨医药(2014年4期)2014-02-27
