
健肝乐颗粒质量评价
2024-03-25 06:13张东方鲁晓光
中成药 2024年3期
张东方,鲁晓光
(南阳市产品质量检验检测中心,河南 南阳 473000)
健肝乐颗粒源于东汉张仲景《伤寒论·太阳篇》,由白芍、甘草组成,为经典名方发展而来的现代制剂。该组方具有养血护肝,解毒止痛的功效。具有降低转氨酶,消褪黄疸以及改善各类肝炎临床症状的作用,临床用于治疗急慢性病毒性肝炎。现执行标准[1]中无含量测定项,无法满足中药现代化的发展需求。目前,该复方制剂研究主要集中在临床方面[2-5],其质量控制研究[6]较少。且目前尚未见有关健肝乐颗粒指纹图谱及化学模式识别的研究报道。在查阅文献[7-14] 的基础上,发现该制剂虽然只有2 味药材,但化学成分多而复杂。本实验以中药指纹图谱结合聚类分析(HCA) 方法、主成分分析法(PCA) 以及正交偏最小二乘判别分析法(OPLS-DA) 将指纹图谱提供的信息进行再评价。再根据不同入血成分保肝作用强弱不同,再经过实验筛选,选取芍药中没食子酸、芍药内酯苷、芍药苷,甘草中甘草苷、芹糖甘草苷、异甘草苷、甘草酸铵作为指标成分,以期为提高健肝乐颗粒的质量标准提供依据,为优化生产工艺达到药效最佳提供基础。
1 材料
Agilent 1260 型高效液相色谱仪(美国Agilent 公司);BP211D 型电子天平(德国赛多利斯公司); WT-1200 型超声波清洗器(济宁万通超声仪器设备厂)。异甘草苷(批号17042102,纯度98%)、芍药内酯苷(批号19121705,纯度99.28%)、芹糖甘草苷(批号20081705,纯度98%)对照品(成都普菲德生物技术有限公司); 芍药苷(批号110736-201539,纯度96.4%)、甘草酸铵 (批号110731-202021,纯度96.2%)、没食子酸 (批号110831-200803,纯度 90.1%)、甘草苷 ( 批号 111610-201908,纯度92.0%) 对照品(中国食品药品检定研究院)。健肝乐颗粒购于武汉康乐药业有限公司,批号1322014、1323014、1322004、1323003、1323002、1322008、1220037、1222036、1222035、1222061、1322013、1322008、1323001、1323002、1220035、1220034,编号S1 ~S16,其中S1 ~S6、S11 ~S14 规格为15 g/袋,含蔗糖; S7 ~S10、S15~S16 规格为6 g/袋,无蔗糖。白芍、甘草购于市场,经南阳市产品质量检验检测中心鲁晓光副主任药师鉴定为正品。甲醇、乙腈均为色谱纯(美国GIOVINI Chemical 公司); 水为超纯水。
2 方法与结果
2.1 色谱条件 Welchrom C18色谱柱(4.6 mm×250 mm,5 μm); 流动相乙腈(A) -0.3%磷酸(B),梯度洗脱(0 ~10 min,10% ~20% A; 10 ~22 min,20% A; 22 ~35 min,20% ~40%A; 35~50 min,40%A); 体积流量1.0 mL/min;柱温30 ℃; 检测波长230 nm; 进样量10 μL。
2.2 溶液制备
2.2.1 对照品溶液 精密称取没食子酸、芍药内酯苷、芍药苷、甘草苷、芹糖甘草苷、异甘草苷、甘草酸铵对照品适量,甲醇溶解并稀释,制成质量浓度分别为25.21、24.71、92.84、11.86、12.69、3.74、46.95 μg/mL 的溶液,即得。
2.2.2 供试品溶液 取本品适量,混匀研细,精密称取0.5 g (S7~S10 各取0.2 g),精密加入25 mL 甲醇,称定质量,超声提取30 min,放冷,甲醇补足减失的质量,摇匀,即得。
2.2.3 阴性样品溶液制备 按处方和工艺,分别制备缺白芍、缺甘草的阴性样品,按 “2.2.2” 项下方法制备,即得。
2.3 方法学考察
2.3.1 精密度试验 取本品(S1) 适量,按“2.2.2” 项下方法制备供试品溶液,在“2.1” 项色谱条件下进样测定6 次,测得各成分相对保留时间、相对峰面积(以峰8甘草苷为参照) RSD 均小于1.9%,表明仪器精密度良好。
2.3.2 重复性试验 取本品(S1) 适量,按“2.2.2” 项下方法平行制备6 份供试品溶液,在“2.1” 项色谱条件下进样测定,测得各成分相对保留时间、相对峰面积(以峰8 为参照) RSD 均小于1.7%,表明该方法重复性良好。
2.3.3 稳定性试验 取本品(S1) 适量,按“2.2.2” 项下方法平行制备6 份供试品溶液,于0、2、4、6、8、12、24 h 在“2.1” 项色谱条件下进样测定,测得各成分相对保留时间、相对峰面积 (以峰8 为参照) RSD 均小于2.0%,表明溶液在24 h 内稳定性良好。
2.4 HPLC 指纹图谱建立
2.4.1 图谱生成 取16 批样品,按“2.2.2” 项下方法制备供试品溶液,在“2.1” 项色谱条件下进样测定,采用“中药色谱指纹图谱相似度评价系统(2012) ”,设置S1为参照,时间窗宽度为0.3 min,生成方法为中位数法,全峰匹配结合多点校正,得到共有指纹图谱和对照指纹图谱(R),见图1A。通过与对照品色谱图(图1B) 比对,确认峰1 为没食子酸,峰4 为芍药内酯苷,峰5 为芍药苷,峰7 为芹糖甘草苷,峰8 为甘草苷,峰15 为异甘草苷,峰20 为甘草酸铵,并选取分离度好、峰面积稳定、保留时间适宜的甘草苷(峰8) 作为参照峰。
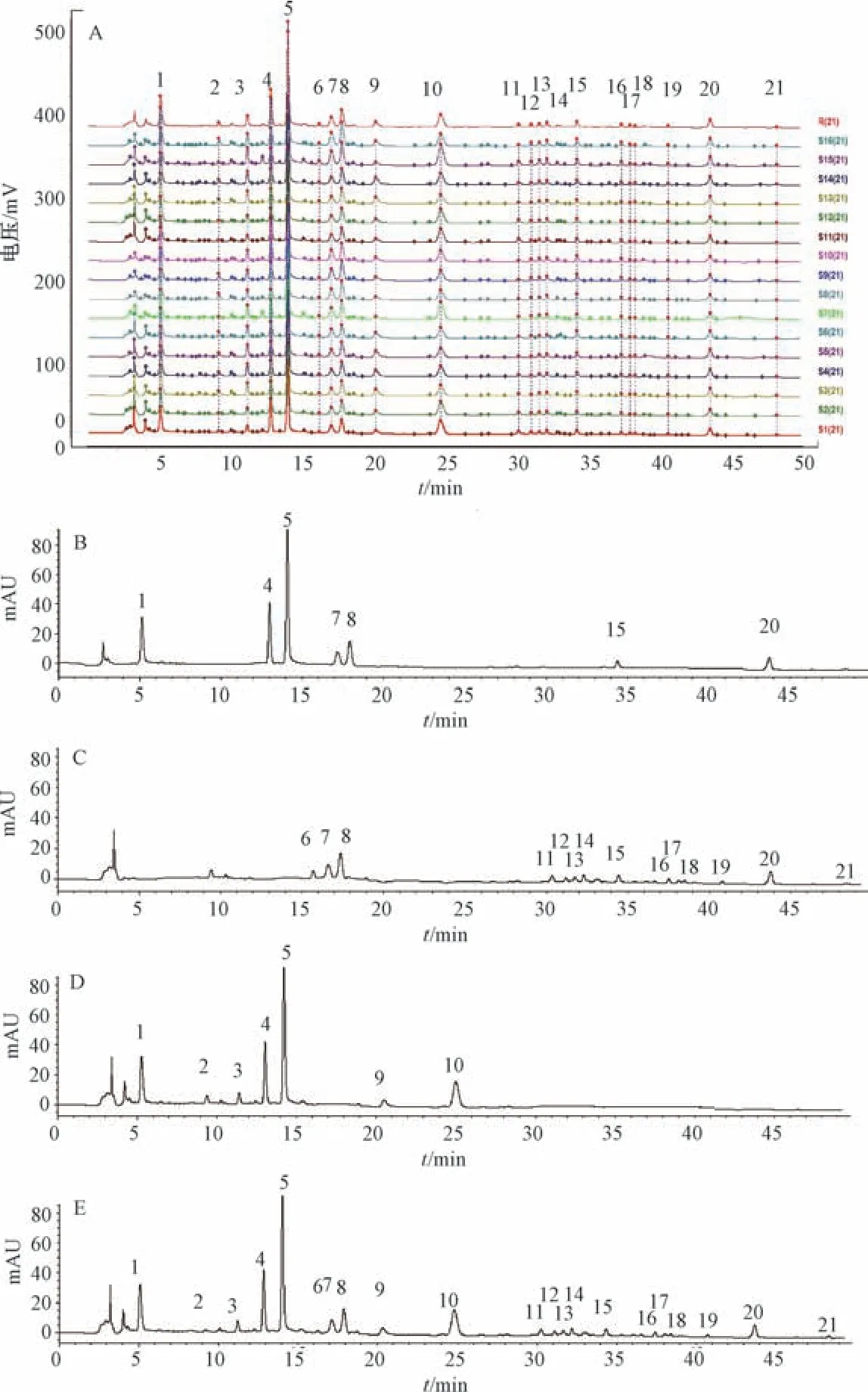
图1 各成分HPLC 色谱图
2.4.2 色谱峰归属 依据单味药材、对照品溶液的紫外光谱信息和保留时间进行比对,结果见图1C~1D。由此可知,色谱峰1~5、9~10 归属于白芍,6~8、11~21 归属于甘草。
2.4.3 相似度评价 16 批样品与对照指纹图谱的相似度分别为0.986、0.985、0.996、0.996、0.995、0.996、0.974、0.992、0.991、0.989、0.985、0.985、0.997、0.997、0.985、0.992,表明不同批次、工艺样品成分组成基本一致,质量均一性良好。
2.5 化学模式识别
2.5.1 聚类分析 (HCA) 采用SPSS 26.0 软件,以Euclidean 距离为度量标准,通过组间对比法,以16 批样品共有峰峰面积与相对峰面积为变量进行系统聚类[15-20],结果见图2。由此可知,以共有峰面积为变量时,16 批样品聚为2 类,S1 ~S6、S8、S9、S11 ~S14、S16 为一类,S7、S10、S15 为一类,表明它们在成分含量上整体一致,但也具有一定的差异性,可能与生产工艺、投料量不同有关,但总体上不大; 以相对峰面积为变量时,16 批样品可聚为2 类,S1~S5、S7~S16 为一类,S6 为一类。综上所述,不同批次、规格、剂型样品成分组成相似度高,质量稳定可靠。

图2 16 批健肝乐颗粒聚类分析图
2.5.2 主成分分析(PCA) 采用SIMCA 14.0 软件对16批样品共有峰峰面积进行PCA 分析,结果见图3A。由此可知,21 个共有峰分成4 个主成分,16 批样品分为2 类,S1~S6、S11~S14 为一类,S7~S10、S15~S16 为一类。

图3 16 批健肝乐颗粒OPLS-DA 分析图
2.5.3 正交偏最小二乘判别分析 (OPLS-DA) 采用SIMCA 14.0 软件对16 批样品进行OPLS-DA 分析,结果见图3。由此可知,VIP 大于1 的成分有11 种,峰号分别为5~9、11、13、15、16、18、21;R2Y为0.992,Q2为0.977,均不小于0.50,表明模型稳定性、预测性良好[19-23]; 经200 次置换检验后Q2回归线与纵轴的相交点小于零,表明模型不存在过拟合,可用于预测分析。
2.5.4 指标成分筛选 中药质量标志物[21](Q-marker) 可反映中药安全性和有效性,并能进行定性定量分析。研究中药质量标志物可提高质量一致性、可控性和溯源性,更好地控制中药生产过程、进行质量监管。本实验根据文献[7-14] 报道及上述结果,确定没食子酸(峰1)、芍药内酯苷(峰4)、芍药苷(峰5)、芹糖甘草苷(峰7)、甘草苷(峰8)、异甘草苷(峰15)、甘草酸铵(峰20) 作为指标成分。
2.6 指标成分含量测定 采用HPLC 法。
2.6.1 溶液制备 同“2.2” 项。
2.6.2 色谱条件 同“2.1” 项。
2.6.3 系统适用性考察和专属性试验 取对照品、供试品(S1)、阴性样品溶液适量,在“2.1” 项色谱条件下进样测定,结果见图1。由此可知,各指标成分分离度符合要求,理论塔板数均大于4 000,阴性无干扰,表明该方法专属性良好。
2.6.4 线性关系考察 分别取对照品溶液1.0、2.0、5.0、10.0、15.0、20.0 μL,在“2.1” 项色谱条件下进样测定。以对照品进样量为横坐标(X),峰面积为纵坐标(Y) 进行回归,结果见表1,可知各指标成分在各自范围内线性关系良好。

表1 各指标成分线性关系
2.6.5 精密度试验 取对照品溶液适量,在“2.1” 项色谱条件下进样测定6 次,测得没食子酸、芍药内酯苷、芍药苷、甘草苷、芹糖甘草苷、异甘草苷、甘草酸铵峰面积RSD 分别为1.8%、1.6%、1.0%、0.41%、0.92%、1.8%、1.2%,表明仪器精密度良好。
2.6.6 稳定性试验 取同一份供试品溶液(S1),于0、2、4、8、12、24 h 在“2.1” 项色谱条件下进样测定,测得没食子酸、芍药内酯苷、芍药苷、甘草苷、芹糖甘草苷、异甘草苷、甘草酸铵峰面积RSD 分别为2.0%、0.82%、1.4%、1.3%、1.1%、2.0%、2.0%,表明溶液在24 h 内稳定性良好。
2.6.7 重复性试验 取供试品溶液(S1) 6 份,在“2.1”项色谱条件下进样测定,测得没食子酸、芍药内酯苷、芍药苷、甘草苷、芹糖甘草苷、异甘草苷、甘草酸铵含量RSD 分别为1.5%、1.4%、0.9%、2.0%、0.9%、0.5%、1.1%,表明该方法重复性良好。
2.6.8 加样回收率试验 精密称取各成分含量已知的样品(S1) 0.25 g,共6 份,加入对照品溶液适量,按“2.2.2”项下方法制备供试品溶液,在“2.1” 项色谱条件下进样测定,计算回收率。结果,没食子酸、芍药内酯苷、芍药苷、甘草苷、芹糖甘草苷、异甘草苷、甘草酸铵平均加样回收率分别为98.41%、97.51%、96.13%、95.59%、96.16%、97.62%、98.09%,RSD 分别为2.0%、1.9%、1.3%、1.6%、1.1%、1.1%、1.1%。
2.6.9 测定方法 取出16 批样品内容物,混匀后研细,按“2.2.2” 项下方法制备供试品溶液,在“2.1” 项色谱条件下进样测定,计算含量,结果见表2。

表2 各指标成分含量测定结果(mg/袋,n=3)
3 讨论
3.1 提取方法选择 先进行了提取溶剂的筛选实验,结果表明在甲醇溶液中,待测成分提取较充分。进一步考察提取方式,分别比较了加热回流30、120、150 min 及超声处理30、40、60、90 min,结果显示甲醇超声处理30 min 时目标成分有效溶出。
3.2 色谱条件考察 考察了不同流动相,包括乙腈-水、甲醇-0.3%磷酸、乙腈-0.3% 磷酸、乙腈-0.3% 甲酸,发现乙腈-0.3%磷酸梯度洗脱时分离效果良好,保留时间适宜,色谱峰峰形佳。通过紫外扫描,发现7 种成分分别在258、230、360 nm 处得到最大吸收,230 nm 处7 种成分均有较强的紫外吸收,且基线最稳定。通过与文献[22-26] 及本实验比较,发现波长切换法和230 nm 单波长测出的结果差异不大,考虑到基线稳定性和重复性,选择230 nm 作为检测波长。
3.3 指纹图谱在复方中药制剂分析中的应用及其作用 中药质量影响因素包括植物种属、采收期、栽培区域、贮藏条件、加工工艺等,进而造成成分差异,使其药理活性及临床应用也存在差异,并使中药质量控制及评价成为复杂难题。指纹图谱能够与中医“整体” 的传统理论呼应,将多指标成分定量法与指纹图谱结合,同时提供定性和定量信息,将是一种更适合、更有吸引力的用于复方中药制剂的分析方法,能更好地揭示不同厂家批次间中药制剂的细微差异。
在指标成分筛选过程中发现,该色谱系统还可同时分离测定芍药、甘草中另外几种具有保肝作用的指标成分丹皮酚、槲皮素、异鼠李素,但在该制剂中含量过低,暂不进行定量研究。中药现代化的发展离不开对有效成分量效关系的深入研究,经典名方尤其应注重多组分的含量研究,以改进生产工艺,使制剂疗效达到最佳。
猜你喜欢
今日农业(2022年13期)2022-09-15
上海文化(文化研究)(2021年6期)2022-01-07
基层中医药(2021年3期)2021-11-22
中老年保健(2021年9期)2021-08-24
中成药(2019年12期)2020-01-04
中国卫生标准管理(2015年4期)2016-01-14
西南医科大学学报(2016年4期)2016-01-03
中国当代医药(2015年33期)2015-03-01
火花(2015年3期)2015-02-27
中国药业(2014年20期)2014-05-17
