
1例由p.Arg35Cys引起遗传性异常纤维蛋白原血症家系的研究
2024-05-07 09:34唐勇张福勇蒋燕珍吴崇荣代洪飞杨红海
右江医学 2024年3期
唐勇 张福勇 蒋燕珍 吴崇荣 代洪飞 杨红海

基金項目:广西壮族自治区卫生健康委员会自筹经费科研课题(Z20200085)
第一作者简介:唐勇,男,副主任技师,医学学士, 研究方向:临床检验诊断学。E-mail:xhero521@163.com
通信作者:杨红海。E-mail:57179627@qq.com
[本文引用格式]唐勇,张福勇,蒋燕珍,等.1例由p.Arg35Cys引起遗传性异常纤维蛋白原血症家系的研究[J].右江医学,2024,52(3):215-220.
【摘要】 目的 分析武鸣壮族地区1例FGA基因杂合突变引起的遗传性异常纤维蛋白原血症的表型和基因型,探讨其发病机制。
方法 对先证者家系三代5人进行外周血采集,使用凝血分析仪检测PT、APTT、FIB、TT等凝血项目,FIB采用PT演算法和Clauss法检测;采用DETA抗凝管收集全血,通过NGS筛选,用Sanger测序验证FGA、FGB、FGG基因编码区突变。
结果 先证者及其父亲表现出FIB-Clauss法水平降低和凝血酶时间延长,而其妹妹和两个女儿均正常。高通量基因测序显示先证者及其父亲检测到FGA c.103C>T杂合错义突变,而先证者的妹妹和两个女儿未检测到该突变。
结论 导致该家系成员遗传性异常纤维蛋白原血症的分子机制是FGA c.103C>T杂合错义突变,该突变导致蛋白质中第35位氨基酸由精氨酸变为半胱氨酸(p.Arg35Cys),从而导致遗传性异常纤维蛋白原血症。
【关键词】 基因突变;家系;遗传性异常纤维蛋白原血症
中图分类号:R446 文献标志码:A DOI:10.3969/j.issn.1003-1383.2024.03.005
A research on 1 case of hereditary abnormal fibrinogenemia family caused by p.Arg35Cys
TANG Yong1, ZHANG Fuyong2, JIANG Yanzhen1, WU Chongrong1, DAI Hongfei1, YANG Honghai1▲
(1. Department of Clinical Laboratory, Nanning Wuming District Hospital of Traditional Chinese
Medicine, Nanning 530199, Guangxi, China; 2. Department of Clinical Laboratory, the First
Affiliated Hospital of Guangxi Medical University, Nanning 530021, Guangxi, China)
【Abstract】 Objective To analyze the phenotype and genotype of 1 case of congenital dysfibrinogenemia caused by heterozygous mutation of the FGA gene in Zhuang region of Wuming, so as to explore the pathogenesis of the disease.
Methods 5 peripheral blood samples from three generations of the proband were collected, and blood coagulation items such as prothrombin (PT), activated partial thromboplastin(APTT), fibrinogen (FIB), and thrombin time (TT) were analyzed by coagulation analyzer, FIB was detected by PT algorithm and Claus method, whole blood was collected by DETA anticoagulant tubes and was screened through NGS, and mutations in the coding regions of the FGA, FGB, and FGG genes were verified by Sanger sequencing.
Results The proband and his father showed reduced levels of the FIB-Clauss method and prolonged time of thrombin, while his sister and 2 daughters were normal. High-throughput gene sequencing revealed a heterozygous for the FGA c.103 C > T missense mutation detected in the proband and his father, which was not found in the detection of his sister and 2 daughters.
Conclusion The molecular mechanism that leads to congenital dysfibrinogenemia in members of this family is FGA c.103C>T heterozygous missense mutation, which causes the 35th amino acid in the protein to change from arginine to cysteine (p.Arg35Cys), resulting in congenital dysfibrinogenemia.
【Keywords】 gene mutation; family; congenital dysfibrinogenemia
遗传性异常纤维蛋白原血症(congenital dysfibrinogenemia, CD)是一种遗传性血液病,由于纤维蛋白原(fibrinogen, FIB)基因缺陷导致FIB分子结构与功能异常,可能引起机体凝血功能异常。CD患者临床表现多样,大部分无症状,少数出现血栓、出血事件或肺动脉高压等症状[1]。CD主要为常染色体显性或共显性遗传,少数为常染色体隐性遗传。由于广泛的临床症状,CD诊断困难[2],治疗方法包括纤维蛋白原替代治疗、抗凝、血栓预防和多学科团队方法[3]。目前尚未建立统一的标准CD治疗方案[2]。本研究发现1例患者纤维蛋白原异常降低,为探明其原因进行家系表型与基因型分析,探讨发病机制,减少误诊漏诊,为该病的研究提供临床依据。
1 对象与方法
1.1 对象
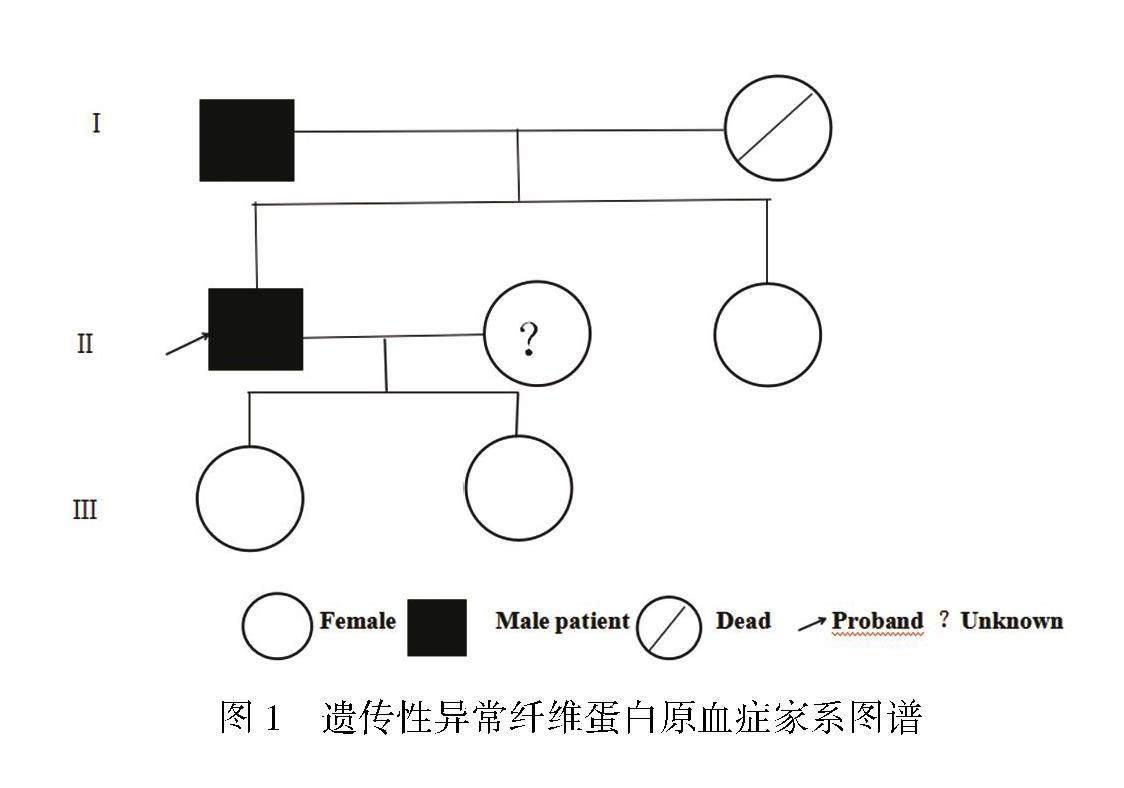
先证者为一名44岁的广西南宁市武鸣区壮族男性。入院体检时发现凝血功能异常,FIB结果为0.47 g/L, TT为40.4 s,PT演算法纤维蛋白原结果正常。其血栓弹力图结果显示凝血因子(R)为5.6 min,纤维蛋白原功能(K)为2.9 min,纤维蛋白原功能(ANGLE)为54.6 deg,血小板功能(MA)为56.8 mm,凝血综合指数(CI)为-1.4,纤溶指标(LY30)为0,纤溶指标(EPL)为0,均在正常范围内。肝肾功能、血常规、D二聚体、纤维蛋白原降解产物(FDP)等结果均未见异常。体格检查结果正常,既往无渗血不止、出血、黑便、尿血或血栓形成病史,也没有家族遗传病史。其家系图谱如图1所示,母亲已逝,其资料不详。
1.2 方法
1.2.1 标本采集
经先证者与家属签署知情同意书后,并通过医院伦理委员会批准,采集先证者及先证者的父亲、妹妹、两个女儿空腹外周静脉血标本各2 mL和4 mL分别置于4支试管。其中1管用枸橼酸钠抗凝,分别进行凝血相关项目检测;另2管用EDTA抗凝进行血常规项目以及基因项目检测,并将该家系的血样本放置于-80 ℃的冰箱中冰冻保存;4 mL促凝管用于生化项目肝功能检测。
1.2.2 凝血项目检测
将收集好的先证者以及家属的凝血标本离心后,按照仪器操作说明书使用法国STA-MAX全自动血凝分析仪及其配套试剂进行凝血相关项目检测(分析仪及其配套试剂需进行质控);SYSMEX CS 2000i全自动血凝分析仪及其配套试剂通过PT演算法测定FIB结果。
1.2.3 血常规检测
将收集好的先证者和家属的血常规标本混合后,按照仪器操作说明书,使用MEK9100全自动血细胞分析仪及其配套试剂进行检测。
1.2.4 肝功能检测
收集好的先证者及其家属促凝管标本离心分离血清后,使用日立LABOSPECT 008AS全自动生化分析仪及其配套试剂对血清标本进行检测。
1.2.5 基因检测
将存放在 -80 ℃ 的血样本送至武汉华中科技大学同济医学院附属同济医院检验科,使用Life Technologies 公司 Ion Torrent PGM 高通量测序仪筛选 FGA、FGB、FGG 基因编码区及其上下游 5bp 范圍内的点突变和微小插入/缺失,并使用 Sanger 测序验证突变。
参考序列:
FGA:GRCh38, Chromosome 4,NC_000004.12, NM_000508.4;
FGB:GRCh38, Chromosome 4,NC_000004.12, NM_005141.4;
FGG:GRCh38, Chromosome 4,NC_000004.12, NM_000509.5。
参考数据库:
The Genome Aggregation Database (gnomAD): http://gnomad.broadinstitute.org/;
Human Gene Mutation Database (HGMD):http://www.hgmd.cf.ac.uk/。
1.2.6 蛋白模型构建
为了描述突变体FIB蛋白的三维结构变化,首先在NCBI上搜索FGA基因的氨基酸序列,将该序列导入SWISS-MODEL生成其野生型分子模型。接着将序列中的第35位氨基酸Arg修改为突变的Cys,再次导入上述工具生成突变型分子模型。最后,使用Swiss-PdbViewer v4.1软件对野生型和突变型的分子蛋白模型进行评估。
2 结 果
2.1 血常规、肝功能、凝血项目
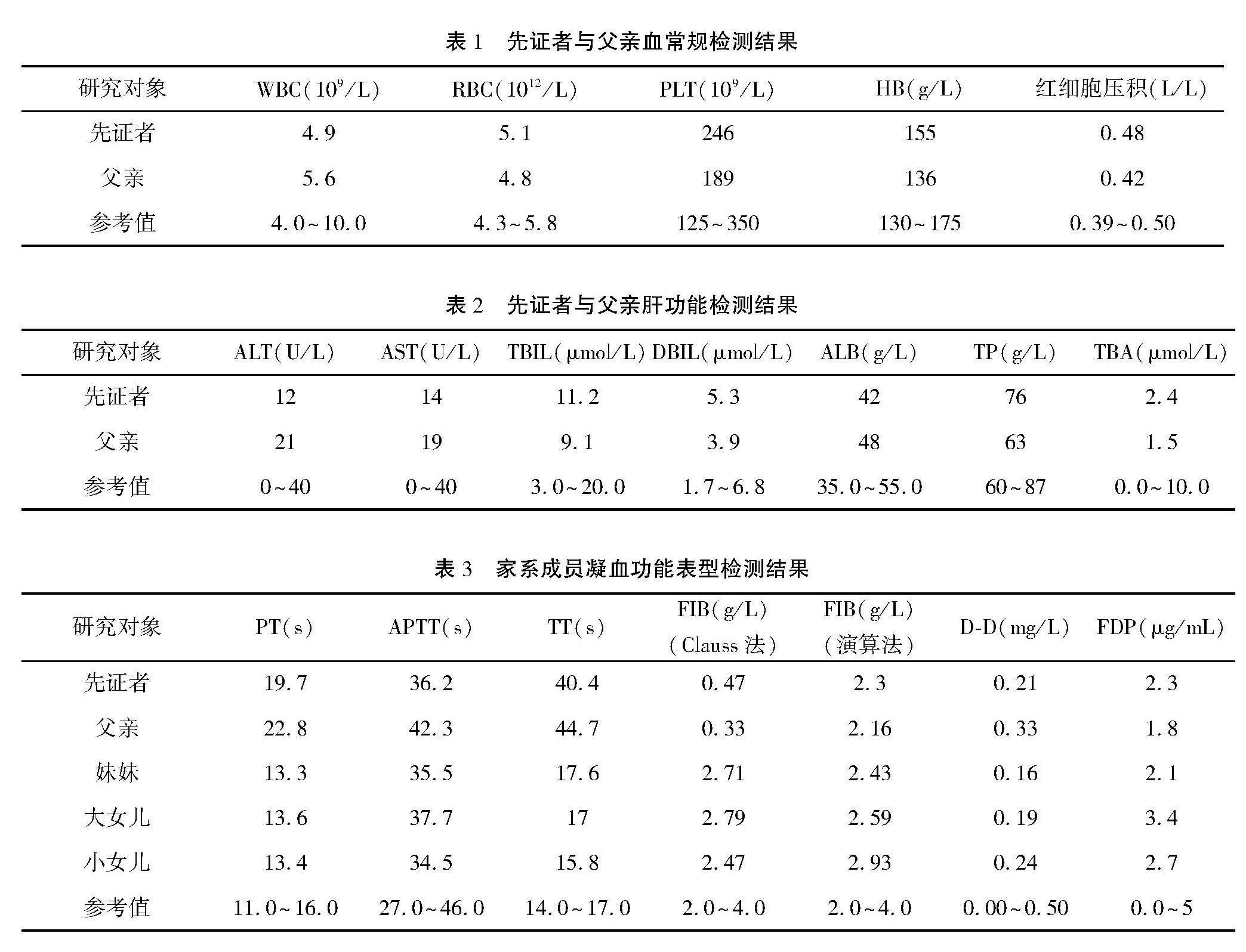
先证者以及家属血常规、肝功能、D二聚体和FDP检查结果均正常,因肝脏疾病引起的FIB缺陷以及纤溶亢进引起的FIB消耗可以排除。先证者的妹妹及女儿凝血功能各项指标均在正常范围内;而先证者与其父亲PT结果均为轻度延长,TT结果延长,FIB-Clauss法结果降低,FIB演算法结果正常。具体结果见表1,表2,表3。
2.2 基因检测结果
先证者及其父亲FIB FGA基因中发现了c.103C>T杂合错义突变。该突变导致蛋白质第35位氨基酸由精氨酸突变为半胱氨酸(p.Arg35Cys)。值得注意的是,先证者的妹妹和女儿的基因结果均为正常,未检测到任何异常突变。据gnomAD数据库显示,该突变在人群中的频率为3.23E-05,在HGMD数据库中也有相关病例报道(CM930249)。综合考虑,该突变被认为具有可能致病性。具体的检测结果见表4,测序结果见图2和图3。
2.3 突变蛋白分子建模
对p.Arg35Cys 突变的分子模型和结构分析,图4A 中白色箭头所指的是野生型 FIB 蛋白 Aα 的第35位氨基酸精氨酸(Arg)。该氨基酸突变为半胱氨酸(Cys)后,蛋白的空间构象发生改变(图4B 红色箭头所指),可能影响凝血酶对纤维蛋白原肽 A 的裂解,导致纤维蛋白肽 A 的释放延长或出现缺陷。
3 讨 论
纤维蛋白原,又称凝血因子Ⅰ,主要由肝细胞分泌[4]。其分子量为340 kD的糖蛋白,能相互结合形成纤维蛋白凝块并介导血小板聚集,在维持机体止凝血平衡方面发挥着重要作用[5]。FIB异常性疾病通常可分为两类:一类是药物引起的获得性异常,另一类是由FIB编码基因缺陷引起的遗传性凝血功能异常疾病[6]。遗传性的FIB异常相对较罕见,发病率约为1/106,可分为两种类型[7]:Ⅰ型为FIB水平降低或缺陷,包括低纤维蛋白原血症和无纤维蛋白原血症;Ⅱ型为FIB分子结构或功能异常,包括低异常纤维蛋白原血症和异常纤维蛋白原血症[8]。CD属于Ⅱ型纤维蛋白原缺陷症。
FIB是由多肽链Aα、Bβ、γ构成的二聚体[9],同源基因FGA、FGB和FGG按顺序编码形成FIB的3条肽链[5],编码基因如出现大片段缺失、框移突变、无义突变、错义突变等缺陷可導致FIB分子结构和功能异常,导致纤维蛋白单体聚合障碍、ⅩⅢ因子介导的交联障碍或纤维蛋白肽A/B释放障碍[10]。本家系先证者在入院体检凝血检查中发现PT 19.7 s,APTT 36.2 s,FIB 0.47 g/L,TT 40.4 s,除FIB活性明显降低、TT延长外,无其他临床症状。基因检测结果显示患者FIB的FGA基因发生c.103C>T杂合错义突变,并导致第35位氨基酸从精氨酸突变成半胱氨酸(p.Arg35Cys)。据研究显示α链上凝血酶裂解位点是R35,错义突变是该点位常见突变之一,从而引起了异常纤维蛋白原血症[11]。从纤维蛋白原基因突变数据库(https://site.geht.org/base-de-donnees-fibrinogene/)也可以证实。此外,该突变可导致纤维蛋白肽A释放延长或缺陷,以及随后的多聚化延迟,最终导致FIB出现异常和TT延长,临床上常无血栓或出血表现[6]。该家系中的先证者临床症状与研究一致。
前期研究表明,女性CD患病率高于男性。这可能是因为女性通常必须接受常规术前检查,如妊娠和产科手术,从而更容易发现凝血功能异常并就诊[9]。本研究对先证者及其父亲、妹妹和女儿进行了相关项目检查。结果显示先证者及家系成员的血常规和肝功能正常,排除了因肝脏疾病引起的FIB缺陷,D-二聚体和FDP也正常,排除了因纤溶亢进导致的FIB消耗。凝血功能结果显示家系中的女儿和妹妹正常,而先证者及其父亲的PT略有延长,TT延长,FIB活性降低。基因检测结果确认仅其父亲的FIB FGA基因c.103C>T存在杂合错义突变,而妹妹和女儿的基因正常,未出现突变。MOSESSON等发现[12],FIB中的Arg变为Cys可增加患者血栓形成的易感性。然而,本研究暂未发现先证者及家系成员出现血栓。但统计数据显示,CD患者发生静脉血栓的中位年龄为34岁,而发生动脉血栓的中位年龄为49岁[13],大出血主要发生在20~40岁[14]。因此,仍需要警惕该先证者发生血栓和出血风险,并需要长期进行血液科随访。FIB基因分析是诊断CD的重要手段,但检出FIB相关基因突变并不等于确诊CD,也可见于遗传性低(无)纤维蛋白原血症。因此,确诊CD尚需结合FIB的Clauss法和PT演算法结果,以及患者的临床表现和家系调查综合诊断。故该病容易漏诊、误诊。特别是仅单独采用Clauss法检测FIB水平常导致该病易被误诊为遗传性低纤维蛋白原血症。一些患者在受到创伤时会引起严重的出血或血栓,甚至危及生命或死亡[15]。通过对该病的认知,有助于提升临床诊疗水平,及时发现并干预,避免误诊和漏诊。
本研究样本来源为受检者血液或体细胞,而非生殖细胞,因此不能排除嵌合现象所致的解读偏差。如果检测结果提示为自发变异位点,该情况下并不排除父母生殖腺嵌合变异的存在。由于人类疾病认识的局限性和本检测Panel所包含基因数量的限制,如未检出FGA、FGB、FGG基因的致病位点,即为阴性结果,但并不能排除患遗传性异常纤维蛋白原血症的可能性。由于对基因认识的不足,对检出的特定基因突变,在某些情况下可能并非引起该疾病的致病突变,尚需要进一步验证和研究。
综上所述,通过本家系的研究,了解其分子发病机制、表型与基因的相关性,为临床对该病的诊断以及治疗提供重要的依据。
参 考 文 献
[1] CASINI A, B LONDON M,LEBRETON A,et al.Natural history of patients with congenital dysfibrinogenemia[J].Blood,2015,125(3):553-561.
[2] JIA Y, ZHANG X W, WU Y S, et al. Congenital dysfibrinogenemia misdiagnosed and inappropriately treated as acute fatty liver in pregnancy: A case report and review of literature[J]. World J Clin Cases, 2022, 10(35): 12996-13005.
[3] LANGER M, MANIRE M, CLARKSON M, et al. Management of congenital dysfibrinogenemia in pregnancy:a challenging patient case[J]. Res Pract Thromb Haemost, 2021, 5(8): e12619.
[4] 韦爱球.FGG基因突变导致遗传性异常纤维蛋白原血症的发病机制研究[D].南宁:广西医科大学,2019.
[5] UNDAS A, CASINI A. Congenital structural and functional fibrinogen disorders:a primer for internists[J]. Pol Arch Intern Med, 2019, 129(12): 913-920.
[6] 刘琳,王卫敏,聂鼎睿,等.1个FGA基因c.103C>T杂合突变导致的遗传性异常纤维蛋白原血症家系分析[J].河南医学研究,2020,6(33):6161-6164.
[7] DE MOERLOOSE P, CASINI A, NEERMAN-ARBEZ M. Congenital fibrinogen disorders:an update[J]. Semin Thromb Hemost, 2013, 39(6): 585-595.
[8] LEBRETON A, CASINI A. Diagnosis of congenital fibrinogen disorders[J]. Ann Biol Clin, 2016, 74(4): 405-412.
[9] 駱娟,段苏容,王华.1例遗传性异常纤维蛋白原血症的家系分析和诊断报告[J].四川大学学报(医学版),2022,53(1):171-174.
[10] 向利群,林发全,程鹏,等.遗传性异常纤维蛋白原血症及其治疗[J].广东医学,2017,38(12):1931-1933.
[11] HANSS M, BIOT F. A database for human fibrinogen variants[J]. Ann N Y Acad Sci, 2001, 936: 89-90.
[12] MOSESSON M W, SIEBENLIST K R, MEH D A. The structure and biological features of fibrinogen and fibrin[J]. Ann N Y Acad Sci, 2001, 936: 11-30.
[13] DE MOERLOOSE P, BOEHLEN F, NEERMAN-ARBEZ M. Fibrinogen and the risk of thrombosis[J]. Semin Thromb Hemost, 2010, 36(1): 7-17.
[14] HAVERKATE F, SAMAMA M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen[J]. Thromb Haemost, 1995, 73(1): 151-161.
[15] PEYVANDI F. Epidemiology and treatment of congenital fibrinogen deficiency[J]. Thromb Res, 2012, 130(Suppl 2): S7-S11.
(收稿日期:2023-05-10 修回日期:2023-09-28)
(编辑:梁明佩)
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30
中国现代医生(2022年19期)2022-11-04
临床输血与检验(2022年3期)2022-06-22
昆明医科大学学报(2022年4期)2022-05-23
昆明医科大学学报(2022年3期)2022-04-19
基层中医药(2021年8期)2021-11-02
中南医学科学杂志(2019年6期)2019-12-05
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
重庆医学(2015年12期)2015-03-05
