
正电子发射断层显像剂[18F]FET前体药物的合成
2011-11-23 05:51刘春艳姜申德孟爱国
合成化学 2011年6期
刘春艳, 姜申德, 孟爱国
(1. 天津大学 药物科学与技术学院,天津 300072;2. 河北联合大学 a. 药学系; b. 附属医院,河北 唐山 063000)
正电子发射断层显像(PET)是近年来迅速发展起来的一种先进的核医学诊断技术,已广泛用于临床上的肿瘤诊断和基础研究中。氨基酸类PET药物与目前临床常规显像药物[18F]FDG{2-脱氧-2-[18F]氟-D-葡萄糖}相比较,对肿瘤组织和炎症部位或其它糖代谢旺盛病灶的特异性高,在脑肿瘤显像方面具有独特的优势,所得图像清晰,准确率高,更容易诊断[1,2],因此,放射性标记的氨基酸已经逐渐成为临床上潜在的PET肿瘤显像剂[3,4]。在氟标记的氨基酸中,O-{2-[18F]氟乙基}-L-氨基酸{[18F]FET}有望成为一种新型PET显像药物用于脑部肿瘤的检测和诊断[5]。
[18F]FET常用的标记方法为亲核取代法。Hamacher小组[6,7]和Wang小组[8]分别合成了[18F]FET前体药物N-(叔丁氧羰基)-O-(2-对甲苯磺酰氧乙基)-L-酪氨酸甲酯(5b)和N-(三苯甲基)-O-(2-对甲苯磺酰氧乙基)-L-酪氨酸叔丁酯;18F与其进行SN2亲核氟化反应,随即水解便可得到[18F]FET。这种方法的优劣主要取决于18F与前体药物进行亲核取代反应的离去基团,离去基团反应活性高有利于提高放化标记率。常用的离去基团有三氟甲磺酰氧基(TfO),甲磺酰氧基(MsO),对甲苯磺酰氧基(TsO),对硝基苯磺酰氧基(NsO),环状硫酰氧基,溴和碘等。其中,TfO是反应活性最好的,现已用于[18F]FDG放化标记的前体药物[9]。
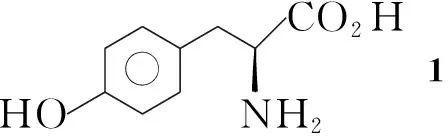


Compa(d)b(e)c(f)RMe(But)Me(But)Me(But)R1SO O - SO O - O2NSO O -
Scheme1
最近,我们报道了以TfO为离去基团,用于[18F]FET放化标记的两个新型前体药物的合成[10,11]。本文选择比TsO反应活性更好的MsO和NsO作为离去基团,采用与文献[10,11]不同的合成路线,以L-酪氨酸为原料,设计并合成了6个[18F]FET前体药物(5a~5f, Scheme 1),其结构经1H NMR, IR, MS及元素分析表征。其中5a,5c~5f未见文献报道。
1 实验部分
1.1 仪器与试剂
Reichert Thermovar型熔点仪(温度未校正);Bruker AV 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Thermo Nicolet Avtar FT 370型傅立叶变换红外光谱仪(KBr压片); EI-MS MS-7070型质谱仪;Flash EA-1112型元素分析仪。
2, 3参考文献[12~14]方法合成;所用试剂均为化学纯;溶剂均作无水处理;所有反应在氮气保护条件下进行。
1.2 合成
(1)N-叔丁氧羰基-O-(2-羟乙基)-L-酪氨酸甲酯(4a)的合成
在反应瓶中依次加入N-叔丁氧羰基-L-酪氨酸甲酯(3a)1.00 g(3.4 mmol), K2CO31.17 g(8.5 mmol), 18-冠-6 0.18 g(0.68 mmol), 四正丁基碘化胺(n-Bu4NI)0.13 g(0.34 mmol)及DMF 25 mL,搅拌使其溶解;滴加氯乙醇0.57 mL(8.5 mmol),滴毕,于125 ℃反应12 h。浓缩后加水30 mL,用二氯甲烷(3×30 mL)萃取,合并萃取液,用无水硫酸钠干燥,浓缩得油状液体4a,直接使用。
用类似的方法合成4d。
(2)5的合成(以5a为例)
在反应瓶中加入4a1.00 g(2.95 mmol)和N,N-二甲基-4-氨基吡啶(DMAP)36 mg(0.295 mmol)及吡啶15 mL,搅拌使其溶解;于0 ℃缓慢加入甲磺酰氯(MsOCl)370 mg(3.24 mmol);于室温反应12 h。浓缩后加水30 mL,用二氯甲烷(3×30 mL)萃取,合并萃取液,用无水硫酸钠干燥, 浓缩呈油状物,经硅胶柱层析[洗脱剂:V(石油醚) ∶V(乙酸乙酯)=3 ∶1]纯化得N-(叔丁氧羰基)-O-(2-甲磺酰氧乙基)-L-酪氨酸甲酯(5a)。
用类似的方法合成5b, 5c。以4d代替4a,同法合成5d~5f。
5a: 无色晶体,收率82%(以3a计算,下同), m.p.71 ℃~73 ℃(乙酸乙酯/石油醚重结晶,下同);1H NMRδ: 7.02(d,J=8.5 Hz, 2H, ArH), 6.80(d,J=8.6 Hz, 2H, ArH), 4.97(d,J=8.1 Hz, 1H, NH), 4.53(dd,J=3.5 Hz, 5.3 Hz, 2H, OCH2), 4.49(m, 1H, CH), 4.18(dd,J=3.5 Hz, 5.3 Hz, 2H, OCH2), 3.69(s, 3H, OCH3), 3.09(s, 3H, CH3), 3.02(m, 2H, CH2), 1.40(s, 9H, CH3in Boc); IRν: 3 380, 2 983, 2 938, 1 733, 1 694, 1 586, 1 511, 1 456, 1 355, 1 246, 1 176 cm-1; MS-EIm/z: 440.2(M++Na); Anal.calcd for C18H27NO8S: C 51.79, H 6.52, N 3.36; found C 52.02, H 6.31, N 3.43。
5b: 乳白色固体,收率80%, m.p.68 ℃~70 ℃;1H NMRδ: 7.82(d,J=8.3 Hz, 2H, ArH), 7.73(d,J=8.3 Hz, 2H, ArH), 7.04(d,J=8.5 Hz, 2H, ArH), 6.87(d,J=8.6 Hz, 2H, ArH), 4.95(d,J=6.7 Hz, 1H, NH), 4.52(m, 1H, CH), 4.35(dd,J=4.0 Hz, 5.5 Hz, 2H, OCH2), 4.11(dd,J=4.1 Hz, 5.4 Hz, 2H, OCH2), 3.71(s, 3H, OCH3), 2.93(m, 2H, CH2), 2.46(s, 3H, CH3), 1.41(s, 9H, CH3in Boc); IRν: 3 385, 2 980, 2 954, 2 927, 1 743, 1 688, 1 597, 1 512, 1 453, 1 362, 1 250, 1 178 cm-1; MS-EIm/z: 516.2(M++Na); Anal.calcd for C24H31NO8S: C 58.40, H 6.33, N 2.84; found C 58.26, H 6.48, N 2.77。
5c: 无色固体,收率76%, m.p.112 ℃~113 ℃;1H NMRδ: 8.37(d,J=8.9 Hz, 2H, ArH), 8.13(d,J=8.9 Hz, 2H, ArH), 6.99(d,J=8.5 Hz, 2H, ArH), 6.65(d,J=8.6 Hz, 2H, ArH), 4.95(d,J=8.7 Hz, 1H, NH), 4.54(m, 1H, CH), 4.50(dd,J=2.5 Hz, 3.8 Hz, 2H, OCH2), 4.15(dd,J=3.7 Hz, 5.3 Hz, 2H, OCH2), 3.71(s, 3H, OCH3), 3.02(m, 2H, CH2), 1.42(s, 9H, CH3in Boc); IRν: 3 377, 2 985, 2 939, 1 744, 1 683, 1 611, 1 536, 1 511, 1 453, 1 368, 1 349, 1 248, 1 123 cm-1; MS-EIm/z: 547.2(M++Na); Anal.calcd for C23H28N2O10S: C 52.66, H 5.38, N 5.34; found C 52.83, H 5.29, N 5.42。
5d: 无色固体,收率81%, m.p.64 ℃~65 ℃;1H NMRδ: 7.08(d,J=7.8 Hz, 2H, ArH), 6.84(d,J=7.8 Hz, 2H, ArH), 4.99(d,J=8.3 Hz, 1H, NH), 4.56(dd,J=4.2 Hz, 6.3 Hz, 2H, OCH2), 4.40(m, 1H, CH), 4.22(dd,J=4.2 Hz, 6.3 Hz, 2H, OCH2), 3.09(s,3H, CH3), 2.98(m, 2H, CH2), 1.40(s, 18H, CH3in Boc); IRν: 3 400, 2 980, 2 934, 1 731, 1 691, 1 615, 1 512, 1 459, 1 354, 1 251, 1 172 cm-1; MS-EIm/z: 482.1(M++Na); Anal.calcd for C21H33NO8S: C 54.88, H 7.24, N 3.05; found C 54.64, H 7.35, N 3.12。
5e: 乳白色油状物,收率83%;1H NMRδ: 7.82(d,J=8.2 Hz, 2H, ArH), 7.36(d,J=8.2 Hz, 2H, ArH), 7.04(d,J=8.6 Hz, 2H, ArH), 6.71(d,J=8.6 Hz, 2H, ArH), 4.96(d,J=6.7 Hz, 1H, NH), 4.39(m, 1H, CH), 4.40(dd,J=4.0 Hz, 5.5 Hz, 2H, OCH2), 4.12(dd,J=4.1 Hz, 5.4 Hz, 2H, OCH2), 2.98(m, 2H, CH2), 2.46(s, 3H, CH3), 1.41(s, 18H, CH3in Boc); IRν: 3 400, 3 007, 2 982, 2 934, 1 731, 1 691, 1 588, 1 509, 1 464, 1 354, 1 252, 1 172 cm-1; MS-EIm/z: 558.3(M++Na); Anal.calcd for C27H37NO8S: C 60.54, H 6.96, N 2.61; found C 60.28, H 7.08, N 2.55。
5f: 无色固体,收率78%, m.p.97 ℃~99 ℃;1H NMRδ: 8.37(d,J=9.0 Hz, 2H, ArH), 8.12(d,J=9.0 Hz, 2H, ArH), 7.04(d,J=8.6 Hz, 2H, ArH), 6.65(d,J=8.6 Hz, 2H, ArH), 4.95(d,J=7.7 Hz, 1H, NH), 4.50(dd,J=5.2 Hz, 3.8 Hz, 2H, OCH2), 4.38(m, 1H, CH), 4.15(dd,J=5.2 Hz, 3.8 Hz, 2H, OCH2), 2.97(m, 2H, CH2), 1.42(s, 18H, CH3in Boc); IRν: 3 399, 2 978, 2 932, 1 736, 1 695, 1 611, 1 537, 1 510, 1 453, 1 366, 1 347, 1 247, 1 181 cm-1; MS-EIm/z: 589.2(M++Na); Anal.calcd for C26H34N2O10S: C 55.11, H 6.05, N 4.94; found C 55.03, H 6.18, N 4.87。
2 结果与讨论
我们按文献[6~8]方法合成5a~5f时,收率较低。为此,设计了Scheme 1的路线合成5a~5f,收率较高。
以L-酪氨酸为原料,在甲醇中缓慢滴加氯化亚砜,即时产生氯化氢催化酯化反应形成甲酯(2a)[12]或在酸催化下与乙酸叔丁酯进行酯交换反应形成叔丁酯(3d)[13]。 2悬浮在含有三乙胺的二氯甲烷中,滴加Boc2O,形成氨基被保护的N-叔丁氧羰基-L-酪氨酸甲酯(3a)或叔丁酯(3d)[14],两步反应的收率分别是81.5%和38.7%。3d收率低是因为第一步酯交换的收率低(2d, 45%)。
在3的酚羟基上引入羟乙基形成关键中间体4。选择氯代乙醇作为羟乙基的来源,3作为亲核试剂,进行SN2亲核取代反应。3只有在碱性环境下形成负离子才能发挥亲核试剂的作用,我们选择碳酸钾为反应体系提供碱性环境[15],加入相转移催化剂18-冠-6增强其在有机溶剂中的碱性,有利于反应进行。氯代乙醇中的氯作为离去基团,其反应活性不强,加入催化剂n-Bu4NI,使反应易于完成。反应在125 ℃,DMF中经12 h完成,经硅胶柱色谱纯化后得晶体4,但收率较低[4a(41%),4d(42%)]。经观察色谱行为,推测4可能在硅胶柱上分解了。实验尝试不经硅胶柱层析直接结晶,但失败了;最后通过快速硅胶短柱除去杂质后结晶的方法,收率较高(68%或71%)。
在吡啶溶液中,以DMAP为催化剂,4a和4d分别与甲磺酰氯(MsOCl),对甲苯磺酰氯(TsOCl),对硝基苯磺酰氯(NsOCl)反应得到5a~5f,收率92%~95%。
实验尝试4的粗品不过柱直接进行下步反应,发现5的收率较高,这进一步说明了4在硅胶柱色谱中分解,可能这类结构的化合物在硅胶柱上不稳定,有待进一步证实。
3 结论
以L-酪氨酸为原料,设计并合成了6个正电子发射断层显像剂[18F]FET前体药物。5a~5c的总收率62%~68%(以3计算),5d~5f的总收率比较低(30%~35%),相关的反应正在改善。目前,这6个化合物的标记和临床研究工作正在进行中。
[1] Couturier O, Luxen A, Chatal J F,etal. Fluorinated tracers for imaging cancer with positron emission tomography[J].Eur J Nucl Med Mol Imaging,2004,31(8):1182-1206.
[2] Laverman P, Boerman O C, Corstens F H,etal. Fluorinated amino acids for tumour imaging with positron emission tomography[J].Eur J Nucl Med Mol Imaging,2002,29(5):681-690.
[3] Fedorova O S, Kuznetsova O F, Shatik S V,etal.18F-labeled tyrosine derivatives:Synthesis and experimental studies on accumulation in tumors and abscesses[J].Bioorg Khim,2009,35(3):334-343.
[4] Iwata R. Development of molecular imaging probes in oncology[J].Gan To Kagaku Ryoho,2008,35(8):1286-1290.
[5] Rickhey M, Koelbl O, Eilles C,etal. A biologically adapted dose-escalation approach,demonstrated for18F-FET-PET in brain tumor[J].Strahlenther Onkol,2008,184(10):536-542.
[6] Hamacher K, Coenen H H. Convenient synthesis of N.C.A.O-{2-[18F]fluoroethyl}-L-tyrosine[J].J Label Compd Radiopharm[J].2001,44:S855-S858.
[7] Hamacher K, Coenen H H. Efficient routine production of the18F-labelled amino acidO-{2-[18F]fluoroethyl}-L-tyrosine[J].Appl Radiat Isot[J].2002,57(6):853-856.
[8] Wang H E, Wu S Y, Chang C W,etal. Evaluation of F-18-labeled amino acid derivatives and [18F]FDG as PET probes in a brain tumor-bearing animal model[J].J Nucl Med Biol[J].2005,32(4):367-375.
[9] Luo L, Tang G, Tang X. Automated synthesis of 2-[18F]-fluoro-2-deoxy-D-glucose by on-column hydrolysis[J].Zhong Nan Da Xue Xue Bao Yi Xue Ban,2009,34(11):1151-1156.
[10] Liu C Y, Jiang S D. Preparation of two triflate precursors forO-{2-[18F] fluoroethyl}-L-tyrosine used in positron emission tomography(PET)[J].Sci China Ser B-Chem[J].2009,52(12):2195-2199.
[11] 刘春艳,姜申德. 正电子发射断层显像剂[18F]FET前体N-叔丁氧羰基-O-(2-三氟甲磺酰氧乙基)-L-酪氨酸甲酯/叔丁酯的合成[J].中国科学B:化学,2010,40(2):189-194.
[12] Aime S, Gianolio E, Corpillo D. Designing novel contrast agents for magnetic resonance imaging.Synthesis and relaxometric characterization of three gadolinium(Ⅲ) complexes based on functionalized pyridine-containing macrocyclic ligands[J].Helv Chim Acta,2003,86(3):615-632.
[13] Taschner Von E, Chimiak A, Bator B,etal. Neue veresterungsmethoden in der peptidehemie ⅧD dar stellung von tert-butylestern freier aminosauren[J].Liebig Ann Chem[J].1961,646(1):134-136.
[14] Kolodziejcyk A M, Manning M. A convenient method forO-alkylation ofN-substituted tyrosines using a crown ether[J].J Org Chem,1981,46(9):1944-1946.
[15] Bender J L, Shen Q D, Fraser C L. Poly(ε-caprolactone) macroligands withβ-diketonate binding sites:Synthesis and coordination chemistry[J].Tetrahedron,2004,60(34):7277-7285.
猜你喜欢
云南化工(2021年9期)2021-12-21
天津医科大学学报(2021年1期)2021-12-05
天然产物研究与开发(2018年1期)2018-02-02
中成药(2018年1期)2018-02-02
现代检验医学杂志(2016年5期)2016-08-20
中国塑料(2016年7期)2016-04-16
海南师范大学学报(自然科学版)(2015年2期)2015-12-23
重庆三峡学院学报(2014年3期)2014-07-16
茶叶通讯(2014年2期)2014-02-27
大学化学(2014年1期)2014-01-26
