
HPLC法测定厄贝沙坦胶囊中厄贝沙坦的含量
2012-08-18 09:38施志顺周志美史学礼王雨艨
中国医药科学 2012年2期
益 磊 施志顺 周志美 李 军 史学礼 王雨艨
安徽省亳州市食品药品检验所,安徽亳州 236800
厄贝沙坦为血管紧张素Ⅱ(AngiotensinⅡ,AngⅡ)受体抑制剂,能抑制AngI转化为AngⅡ,能特异性地拮抗血管紧张素转换酶受体(AT1),对AT1的拮抗作用大于AT28 500倍,通过选择性地阻断AngⅡ与AT1受体的结合,抑制血管收缩和醛固酮的释放,产生降压作用。
厄贝沙坦胶囊为已有国家药品标准的药品,其标准收载于国家药品标准《新药转正标准》第62册,标准号为WS1-(X-026)-2005Z。为了使本产品质量达到可控,参照国家药品标准WS1-(X-026)-2005Z,对本品的含量进行了研究。
1 仪器与试药
1.1 仪器
岛津LC-10ATvp高效液相色谱仪(苏州岛津公司);岛津SPD-10Avp紫外检测器(苏州岛津公司);AB135-S电子天平(d=0.01 mg)(METTLER TOLEDO公 司);Kromasil C18柱(150 mm×4.6 mm, 5μm,日本岛津公司)。
1.2 试药
厄贝沙坦对照品 (批号:100607-200301),由中国药品生物制品检定所提供;厄贝沙坦胶囊(批号:110901、110902、110903);乙腈HPLC(美国TEDIA公司);磷酸 (汕头西陇化工厂);三乙胺(海金山亭新化工试剂厂)
2 方法与结果
色谱条件:色谱柱shimpack VP-ODS(150 mm×4.6 mm,5μm);流动相:磷酸溶液(取85%磷酸5.5 mL,加水至950 mL,以三乙胺调节pH至3.2)-乙腈(62∶38);检测波长: 245 nm;进样量:20 μL ;流速:1.0 mL/min。
2.1 阴性干扰试验
取处方量的辅料,按制备工艺操作制成模拟胶囊。精密称取上述空白辅料适量,按含量测定方法配制供测液。精密量取20μL注入液相色谱仪,记录色谱图。结果表明,阴性不干扰本法的测定。见图1 ~ 3。
2.2 系统适用性试验
取供试品溶液,连续注样10次,分别记录每次主峰面积、保留时间、拖尾因子、理论板数。由系统适用性图谱可看出,本品理论板数按厄贝沙坦计算为7 371.236,拖尾因子为1.027。符合中国药典2010年版二部附录V D的要求,系统评价图谱见图4。

图1 辅料干扰性试验—阴性图谱

图2 辅料干扰性—样品图谱
图3 辅料干扰性—对照图谱
图4 系统适应性—系统评价图谱
2.3 标准曲线线性范围的考察
精密称得厄贝沙坦工作对照品25.05 mg(标定纯度为100.6%),置50 mL量瓶中,加流动相溶解并稀释至刻度,精密量取上述溶液 1、2、3、4、5、6、7 mL,分别置 10 mL 量瓶中,加流动相稀释至刻度,摇匀,取此溶液,照正文含量测定项下色谱条件与方法注样,各浓度注样2针,取峰面积平均值。以峰面积为纵坐标,浓度(mg/mL)为横坐标,分别进行线性回归。计算回归方程、相关系数(R)、截距与标示浓度响应值百分比(B/Y%)结果见表1,线性结果见图5。
回归方程为A=259 675 62.358C-42853.143 (R=0.999 96,n=7)。且截距与限度浓度响应值的比值为-0.83%,曲线通过原点。结果表明,在0.050 4~0.352 8 mg/mL浓度范围内,本法的线性良好。
2.4 回收率
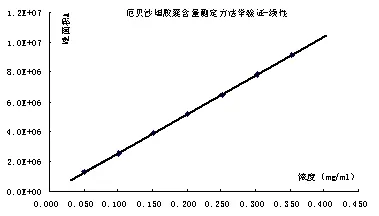
图5 厄贝沙坦含量测定线性图
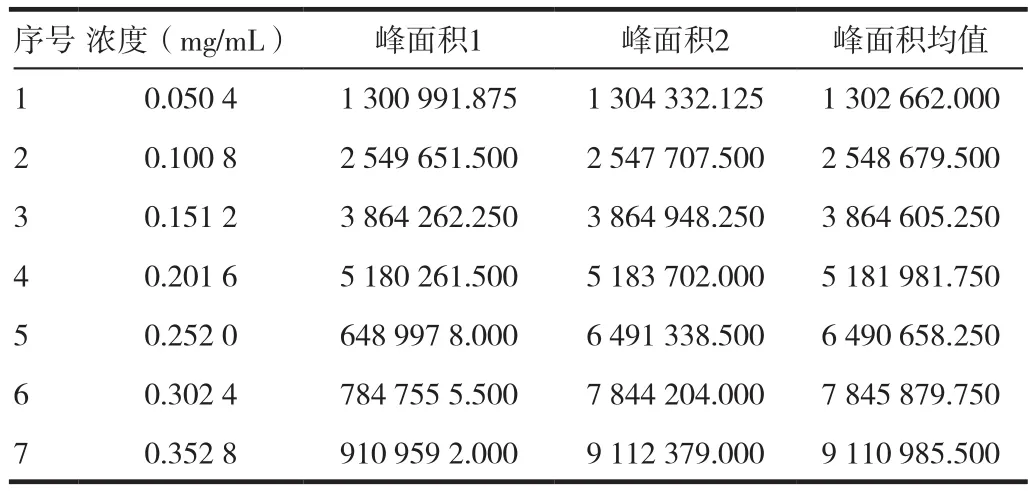
表1 厄贝沙坦含量测定线性试验结果
取厄贝沙坦对照品8、10、12 mg,各3份,精密称定,分别置50 mL量瓶中,加入处方比例的阴性样品,加流动相适量,振摇使厄贝沙坦溶解,用流动相稀释至刻度,摇匀,滤过,精密量取续滤液10μL注入液相色谱仪,记录色谱图;另取厄贝沙坦对照品10 mg,精密称定,置50 mL量瓶中,加流动相溶解并稀释至刻度,作为对照品溶液。同法测定。按外标法以峰面积计算本法的回收率。结果表明,本法测定厄贝沙坦含量的回收率高,9份样品平均回收率均值在98.0~102.0%,RSD<2.0%。见表2。

表2 厄贝沙坦含量测定方法回收率试验结果
2.5 重复性
取同批均质样品(批号:110903)装量差异项下的内容物,研细,精密称取适量(约相当于厄贝沙坦10 mg),共10份,分别置50 mL量瓶中,加流动相适量,振摇使厄贝沙坦溶解,用流动相稀释至刻度,摇匀,滤过,精密量取续滤液10μL注入液相色谱仪,记录色谱图;另取厄贝沙坦对照品10 mg,精密称定,置50 mL量瓶中,加流动相溶解并稀释至刻度,摇匀,作为对照品溶液。同法测定。按外标法以峰面积计算供试品中厄贝沙坦的含量。以含量计算相对标准偏差RSD。结果表明,本品测定厄贝沙坦胶囊含量方法的重复性较好。见表3。

表3 厄贝沙坦含量测定重复性试验
2.6 中间精密度
取同批均质样品10份,在另一色谱系统上测定其含量,考察本法的中间精密度。结果表明,本法测定厄贝沙坦含量中间精密度良好。见表4。

表4 含量测定方法验证—中间精密度数据
2.7 溶液稳定性试验
取重复性项下的同一对照溶液与供试品溶液分别于2、4、6、24、48、72、96 h 各注样 20μL,计算 RSD 值,考察日内与日间溶液的稳定性。结果表明,本法测定厄贝沙坦含量,对照溶液与供试品溶液的日内与日间均较稳定。见表5。

表5 含量测定方法验证—溶液稳定性
3 含量测定
3.1 色谱条件
色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂,以磷酸溶液(取85%磷酸5.5 mL,加水至950 mL,以三乙胺调节pH至3.2)-乙腈(62∶38)为流动相,检测波长为245 nm。理论板数按厄贝沙坦峰计算应不低于2 000。
3.2 测定法
取装量差异项下的内容物,研细,精密称取适量(约相当于厄贝沙坦10 mg),置50 mL量瓶中,加流动相适量,振摇使厄贝沙坦溶解,用流动相稀释至刻度,摇匀,滤过,精密量取续滤液10μL注入液相色谱仪,记录色谱图;另取厄贝沙坦对照品,同法测定。按外标法以峰面积计算,即得。
取本品3批样品,照上述方法进行试验。结果表明,110901、110902及110903三批样品的含量符合要求,分别为99.5%、99.7%和100.1%。
4 讨论
该项参照国家药品标准WS1-(X-026)-2005Z厄贝沙坦胶囊[含量测定]项制定。并进行了方法学验证,结果表明:本法专属性良好,阴性不干扰检测,系统适用性良好,厄贝沙坦在0.050 4~0.352 8 mg/mL的浓度范围内的线性相关系数大于0.999,且截距与限度浓度响应值的比值小于2.0%,曲线通过原点,可使用外标一点法进行计算。回归方程如下:A=25 967 562.358C-42 853.143 (r=0.999 96,n=7)。回收率为99.7%,重复性、中间精密度良好。供试品与对照品溶液在日内与日间均较稳定。故本法可用于本品的厄贝沙坦的含量测定。
[1]熊思敏,吕竹芬,谢清春,等.高效液相色谱法测定厄贝沙坦缓释片的含量[J].中国药师,2008,7(2) :85-87.
[2]朱娟,尤时龙,孟小弟. 复方厄贝沙坦片中厄贝沙坦和氢氯噻嗪的HPLC测定 [J].中国医药工业杂志,2003,34(11):33-35.
[3]刘娇艳,李典赛,陈晓辉. 氢氯噻嗪分散片中厄贝沙坦和氢氯噻嗪的含量[J].亚太传统医药 .2011,8(2):24-26.
[4]孙启泉,刘燕,施介华,等. RP-HPLC法测定厄贝沙坦的含量及有关物质[J].浙江化工, 2006,37(6):28-30.
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
铁军·国防(2022年4期)2022-05-14
南洋资料译丛(2021年1期)2021-11-26
药品评价(2021年17期)2021-11-06
铁军·国防(2021年5期)2021-09-10
中华养生保健(2020年10期)2021-01-18
化工管理(2020年18期)2020-07-15
中国医药指南(2017年3期)2017-11-13
转化医学电子杂志(2015年4期)2015-12-27
中国生化药物杂志(2015年4期)2015-07-07
