
环保型三价铬电沉积功能性铬层研究现状及发展
2013-06-14 06:57冯忠宝屠振密胡会利
电镀与涂饰 2013年3期
冯忠宝,屠振密,胡会利*
(哈尔滨工业大学(威海)应用化学系,山东 威海 264209)
铬镀层由于具有高硬度、高耐磨性和优良的装饰性而得到广泛的应用,因传统镀铬工业中的六价铬毒性大[1],人们期望能研制出环境友好的三价铬电沉积工艺。与六价铬镀铬相比,三价铬电沉积具有很多优良的特性[2]:镀液的覆盖能力和分散能力高,电流效率高,不受断电的影响等。三价铬电沉积铬主要分为硫酸盐型和氯化物型。氯化物体系导电性好,槽压低,电流
效率较高,阳极采用石墨,成本低,初始经济性好,但阳极有氯气析出,对环境有污染,对设备腐蚀较严重。该工艺前期研究较多,随着对环保意识的增强,硫酸盐体系[3]因阳极无氯气析出,不会腐蚀设备,故更有利于环境保护。环保镀装饰铬工艺已较成熟,现已投入生产。
近年来,环保镀功能铬的研究发展很快,已取得较大进展[4],但还存在不少问题,如镀液在长期电沉积中的稳定性、电沉积速率和镀层增厚等,目前仍处于实验室研制阶段,仅能在有限条件下得到有限的厚度,故未能达到批量和工业化生产规模。本文综述了三价铬体系电沉积厚铬工艺的研究进展。
1 环保型电沉积功能铬的几种典型工艺
近10 多年来,环保型电沉积功能性铬工艺研究发展很快,相关的文献报道已超过百篇,现介绍几种典型的环保铬工艺,见表1。
工艺1 连续电沉积30 min 所得镀层平均厚度为92 μm,表面光亮并含有微裂纹,XRD 检测表明铬层为非晶态。
工艺2 的镀液中含400 g/L 二甲基甲酰胺(DMF),在14 A/dm2的电流密度下电沉积10 min 时电流效率在30%以上,随时间延长,电流效率逐渐降低,电沉积60 min 得到镀层的平均厚度为36 μm。
工艺3 在15 A/dm2的电流密度下电沉积60 min 可获得超过30 μm 的铬镀层,维氏硬度达706 HV。经过200 °C 热处理后硬度可提高到1 401 HV。
工艺4 在较低浓度的Cr(III)镀液中进行电沉积,当pH 为1.8 和阴极电流密度为20 A/dm2时持续电沉积2 h,镀层厚度达23.6 μm,硬度为733 HV,镀层与基体结合力良好。
在工艺5 所示基础镀液中加入肼或磷酸羟胺后可得到10 μm 厚的镀层。当电流密度为20 A/dm2时,其表面粗糙度Ra分别为2.229 2、1.453 1 和0.780 1 μm,镀液中加入肼或磷酸羟胺都可使镀层表面光滑,镀层耐蚀性变好。
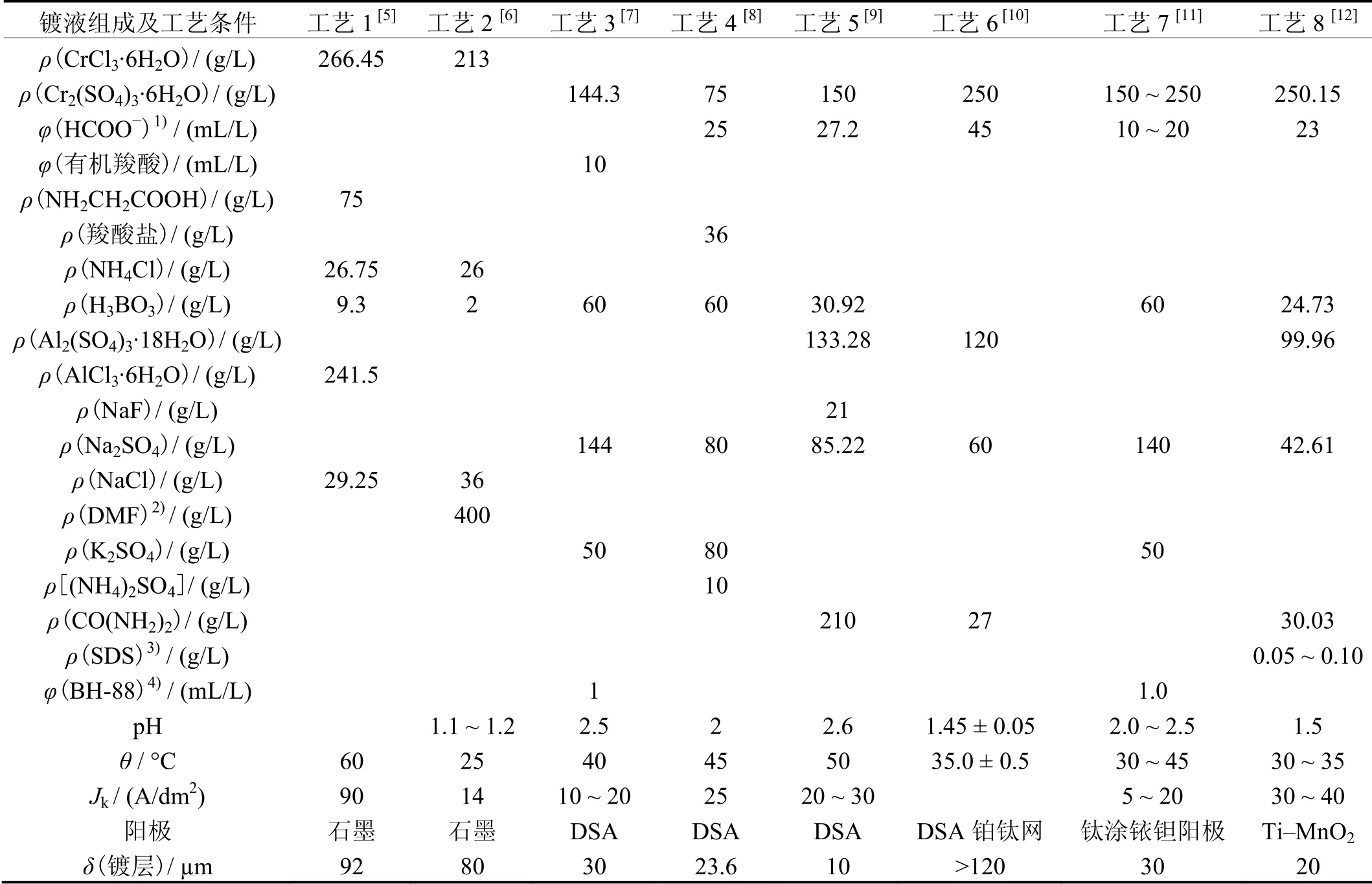
表1 氯化物和硫酸盐体系电沉积功能铬工艺参数Table 1 Process parameters of functional chromium coating from chloride or sulfate system
在工艺6 的高浓度Cr(III)条件下加入3 种配位剂后,三价铬离子放电变为二价铬离子的过程属于扩散控制,二价铬放电变为铬原子受传质和传荷的共同控制,因此,金属铬的沉积属于混合控制。在搅拌条件下连续电沉积9 h,可得到180 μm 厚的镀层。
工艺7 在电流密度为9 A/dm2下电沉积60 min 可得到30 μm 厚的铬镀层,镀层硬度随热处理温度升高而增大,500 °C 时达到1 090 HV,镀层在高于293 °C时转变为晶态。
工艺8 所得镀层硬度为850~950 HV,热处理后硬度提高到1 200~1 300 HV,铬晶粒尺寸3~5 nm,阴极电流效率30%~40%。
上述8 种电沉积三价铬工艺的阴极电流效率较高,沉积速率较快,都能得到较厚的镀层。
2 影响环保铬电沉积的因素及对策
2.1 环保铬电沉积的阴极特征
1981年Z.M.Tu 等[13-14]对氯化物-甲酸盐-乙酸盐三价铬电沉积阴极过程特征进行了研究,用微锑电极测试了阴极表面“电沉积时间与pH(指离阴极表面附近20 μm 处的pH)”及“电沉积时间与电流效率”的关系,结果见图1和图2[15]。

图1 电沉积时间与阴极表面附近pH 的关系Figure 1 Relationship between pH close to cathode surface and plating time
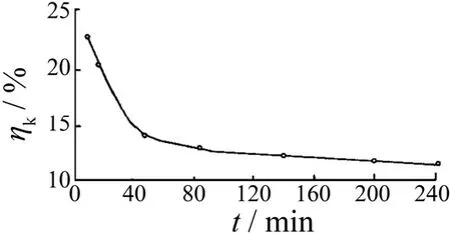
图2 电沉积时间与电流效率的关系Figure 2 Relationship between current efficiency and plating time
Cr3+还原成金属铬的标准电极电势为-0.740 V,由于其电势较负,电沉积时阴极上大量析氢,使阴极表面pH 迅速升高。图1表明随电镀时间延长,pH 迅速升高。pH 高于7 时,开始有Cr(III)羟桥化合物或低聚物生成和积累[16];pH 高于8 时,Cr(OH)3胶体生成并吸附在阴极表面,使镀层质量恶化,电流效率逐渐下降(见图2),电沉积难以继续进行,镀层增厚受阻。
在三价铬电沉积中选择适宜的组合配位剂,采用较高主盐浓度,较低pH 和适宜的缓冲剂,并强化搅拌条件,使镀液维持在较低且稳定的pH 范围内,可有效改善环保镀铬液的稳定性并提高阴极电流效率及镀层厚度。
2.2 影响环保电沉积功能铬的主要因素及对策
2.2.1 配位剂的影响
在不含配位剂的Cr(III)水溶液中是不能电沉积出铬层的,这是由于三价铬离子常以稳定的规则八面体结构[Cr(H2O)6]3+形式存在,这种结构很难使Cr(III)在阴极上得电子,因此铬很难在水溶液中单独沉积析出,必须加入适宜的配位剂,以形成容易电沉积的电化学活性配体,并具有适宜的热力学稳定性[17]。配位剂还能抑制铬的羟桥化反应,提高镀液稳定性[18]。A.Baral等[19]和V.A.Safonov 等[20]认为羧基化合物能起到配位的作用,常用配位剂主要有甲酸及其盐,乙酸及其盐、草酸、尿素、氨基乙酸、柠檬酸和酒石酸等。
研究表明甲酸根是最适宜的配位剂之一,其在水溶液中能置换出[Cr(H2O)6]3+中心的水分子,形成具有一定活性的[Cr(H2O)5CHOO]2+配合离子[21-22]。同时HCOO-也是三价铬镀液良好的稳定剂和抗氧化剂(抑制六价铬的生成),R.Giovanardi 等[23]的研究也证明了这一点。
M.El-Sharif 等[24-26]重点研究了甲酸和甲醇在硫酸盐镀液中的作用。甲酸的主要作用是和三价铬形成配位离子,并能催化三价铬配体的交换;甲醇的作用是可降低阴极析氢量,抑制阴极扩散层的pH 上升,还可与三价铬形成配位化合物,抑制羟基配体和聚合物的形成。镀液中加入甲酸或甲醇后,能明显增大沉积速率和镀层厚度,同时改善镀层质量,提高镀液稳定性,采用该工艺可获得厚度超过200 μm 的铬沉积层。Z.X.Zeng 等[27]研制了分别以甲酸、草酸及氨基乙酸为配位剂的氯化物镀液,就沉积速率而言,以草酸或甘氨酸为配位剂的镀液体系较甲酸沉积速率快,这可能与配位离子的几何结构有关。与镀装饰性铬相比,硬铬的电流效率从0.7%提高到22.2%,粗糙度由0.30 μm变为5.54 μm。
G.Hong 等[28]研究表明,组合配位剂的使用和选择对镀层增厚有较大的影响,采用2 种羧酸配位剂比单一配位剂好,使用3 种羧酸配位剂的综合效果更好,能有效提高沉积速率和沉积层厚度。该工艺使用的三价铬电沉积工艺中加入3 种羧基酸类配位剂,可连续电镀9 h,电流效率达30%~40%,镀层厚度为180 µm;还可高速电沉积20 h,镀层厚度达450 μm;镀层经热处理后,其硬度高达1 200 HV。
研究表明,要在三价铬溶液中电沉积得到功能性铬层,甲酸根是首选的配位剂[21]和稳定剂,还必须选择加入2 种或2 种以上的辅助配位剂(多为羧酸或羧酸盐),形成适宜的组合配位剂,这样才能有效地保持镀液的稳定性,并提高阴极电流效率和沉积速率,从而实现在较长时间内高速电沉积,这是镀层增厚的关键。
在三价铬溶液中加入适宜含碳的有机配位剂,就可得到Cr-C 合金镀层[29-33],该类合金具有很高的硬度和耐磨性以及优良的耐高温性能,镀厚性也很好,厚度可超过60 μm,镀态硬度为600 HV,热处理后硬度可达1 400 HV,该合金很有希望成为功能铬的替代层之一。
2.2.2 主盐浓度的影响
镀液中三价铬的浓度对提高极限电流密度,保持较高的沉积速率和镀层增厚的影响较大。一般而言,随主盐浓度升高,金属离子沉积速率加快,电流效率升高,这是由于Cr(III)配位离子浓度增大可提高扩散速率,并产生更多的电活性Cr(III)配位离子,从而保持高速电沉积,故电沉积厚铬时常使用较高的主盐浓度。高浓度Cr(III)虽然有利于高速沉积,但也受溶解度和溶液黏度增大的限制,通常主盐浓度在0.6~0.8 mol/L 范围内较为合适,不宜过高。
V.S.Protsenko等[34]对以0.5 mol/L甲酸和0.5 mol/L尿素为配位剂的硫酸盐体系的研究结果表明,在15~25 A/dm2的电流密度范围内,电流效率随主盐浓度升高[Cr(III)从0.8 mol/L 增至1.0 mol/L]而增大;但电流密度为15 A/dm2和20 A/dm2时,电流效率增幅比较缓慢;电流密度为25 A/dm2时,电流效率迅速增大。其原因是,主盐浓度的增大能够提高Cr(III)离子的放电几率,从而提高了电流效率。而随电流密度的增大,阴极析氢电位负移,有利于金属铬的沉积,故电流效率随主盐浓度的升高而迅速增长。
王华等[7]指出镀层沉积速率随主盐浓度的升高而增大,但主盐浓度过大时,电沉积时会析出粗大的Cr晶粒而形成黑色硬质条纹,因此,主盐浓度不宜过高。此外还要注意主盐浓度和配位剂的比例。
S.Duan 等[35]在高浓度(0.8~1.4 mol/L)的Cr(III)溶液中加入甲酸和尿素(或草酸)、导电盐、硼酸等,在较高电流密度(20~25 A/dm2)和低pH 下,电流效率可达25%~30%,镀层厚度达70 µm。
2.2.3 镀液pH 和缓冲剂的影响
2.2.3.1 pH 的影响
由图1、2 可知,pH 对镀液的沉积速率和镀厚性的影响很大,pH 过高时会在阴极表面生成羟桥化合物和Cr(OH)3胶体,阻抑沉积层继续增厚。李惠东等[36]和Z.X.Zeng 等[37]的研究结果也证明了这一点,因此三价铬镀层增厚较为困难。
G.Hong 等[28]指出电沉积过程中阴极表面(特别是双电层附近)pH 的上升,会使沉积速率迅速下降,甚至降为零。使用缓冲剂能够调节镀液pH 的变化,防止氢氧化物的形成,对维持镀液成分和沉积速率恒定有一定作用。
Protsenko 等[38]研究了镀厚铬中pH 对电流效率的影响。当pH 在1.2~1.8 范围内时,随pH 的增大,电流效率增大,沉积速率加快。pH 为1.8 时,在35 A/dm2的电流密度下15 min 内的沉积速率达2.26 μm/min,电流效率达43%。当pH 降为1.2 时,由于阴极大量析氢,电流效率下降为15%,沉积速率下降为0.76 μm/min。曾志翔等[39]的研究结果也说明了这一点。
F.I.Danilov 等[12]在pH 为1.5 的高浓度三价铬溶液中得到了纳米铬镀层。可见低pH 有利于形成纳米尺寸的晶粒,且很少有Cr(OH)3的夹杂,从而使镀层硬度提高。
在较长时间的功能铬电沉积中,保持和控制镀液中阴极表面pH 的稳定性非常重要,特别是在阴极扩散层内,pH 直接影响镀液中的Cr(III)与配位剂的配合状态及稳定性。采用较低的pH 有利于提高阴极电流效率、沉积速率和沉积层厚度。
功能性电沉积铬镀液的适宜pH 为1.5~3.0,当pH大于3 时,由于阴极表面羟桥化合物的生成和积累,电流效率和沉积速率降低,并有黑色产物生成;若pH低于1,阴极析氢严重,电流效率降低,沉积速率缓慢,也不适于功能性铬电沉积。
2.2.3.2 缓冲剂的影响
在功能电沉积铬中,一般需要较长时间才能得到较厚的铬镀层,这就需要在电沉积过程中保持和控制镀液(特别是阴极扩散层内)pH 的稳定。随电沉积时间延长,阴极表面的pH 迅速升高,由于羟桥化合物的生成和积累,沉积速率迅速降低;继续升高pH,甚至会生成Cr(OH)3沉淀,铬的沉积完全停止,镀层表面粗糙发暗。
为使镀液中的pH 保持相对稳定,通常需加入适宜的缓冲剂。大量试验表明,硼酸是三价铬电沉积中首选的优良缓冲剂。为了进一步提高镀液的缓冲能力,还可加入适量的铝盐,如氯化铝、硫酸铝等。此外还可加入第3 种缓冲剂,主要是羧酸类化合物,其效果更好[28,40]。
2.2.4 调制电源的作用及应用
调制电源对镀层的性能及厚度均有较大的影响,通过脉冲电沉积可以很好地消除浓差极化,减小阴极表面与本体溶液中铬离子的浓度差,提高镀层沉积速率,改善镀层质量。因此可以采用脉冲技术在三价铬体系中镀功能铬。许多文献报道[41-44]采用脉冲技术可得到纳米晶铬镀层,能明显提高镀层硬度、耐磨性、耐蚀性和厚度,可与六价镀硬铬相媲美,非常适用于环保型功能铬电沉积。
G.Saravanan 等[42]比较了分别采用直流和脉冲电沉积所得铬镀层的耐蚀性。结果表明,脉冲镀铬层表面比较平滑,孔隙率也较小且耐蚀性优良。H.Feng 等[45]的研究也得到同样的结果,他们还采用双向脉冲技术制得纳米晶多层结构铬。
X.K.He 等[43]采用较低频率(50 Hz)脉冲电沉积制备三价铬镀层。结果表明,主盐浓度(2.0 mol/L)和平均电流密度(6 A/dm2)相同时,脉冲镀和直流镀所得镀层厚度分别为11.2 μm 和7.1 μm。脉冲电沉积速率高于直流电沉积,镀层也更厚。
Y.B.Song 等[44]以甲酸铵为配位剂,采用氯化物镀液,在平均电流密度为10 A/dm2、占空比和频率分别为50%和100 Hz 下电沉积相同时间,所得直流和脉冲镀层的厚度分别为2.0 μm 和8.5 μm。这就说明采用脉冲电沉积技术可提高镀层厚度。研究还表明增大脉冲频率,能显著提高镀层的显微硬度,并有效降低镀层内应力。
2.2.5 其他因素
由于三价铬功能性电沉积过程的机理还不够清楚,镀液成分和工艺条件多而复杂,影响电沉积增厚的因素很多,除对以上几种主要因素加以控制外,采用双槽或用离子交换膜将阴、阳极分开,加强搅拌(磁力或超声波),高速流动镀液等方法,也能有效地提高阴极电流效率、沉积速率和沉积层厚度。
3 结论与展望
上世纪70年代,环保铬电沉积仅能得到3~4 µm的铬镀层,只能用作装饰性镀层。近30年来,随Cr(VI)应用的严格控制及工业现代化的需要,环保型电沉积功能铬得到迅速发展,已经取得了相当大的进展。目前在较高浓度三价铬溶液中,加入优选的组合配位剂,采用较高的阴极电流密度和较低的pH,能够得到一定厚度的功能性铬层。但目前存在的问题和困难还比较多,如电沉积较长时间时,镀液的稳定性还不够好,且随电沉积时间延长,阴极电流效率下降明显、沉积速率减缓,镀层质量下降严重。
目前对电沉积功能铬的研究,基本上是通过大量试验和摸索,缺乏重要理论的有力指导,从而影响电沉积功能铬的进一步发展,这也是功能铬发展的瓶颈。电沉积功能性铬层要取得突破,应大力开展对环保铬电沉积机理的研究,主要从以下3 个方面入手:
(1) 加强环保铬配位机理的研究。由于三价铬与水之间能形成很稳定的配合物,需要加入其他配位剂以降低三价铬配离子的放电难度。当多种配位剂同时存在时,三价铬的配位状态比较复杂,有必要采用某些分析方法研究三价铬离子与组合配位剂形成的配合物结构,同时需要研究pH、温度等诸多因素对配合物结构及稳定性的影响。
(2) 加强环保铬电化学还原机理的研究。至今对三价铬阴极还原的历程还不十分清楚:三价铬还原所得的3 个电子是分3 步进行、每次得1 个电子,还是先得1 个电子再得2 个电子,或是先得2 个电子后再得1 个电子;这些步骤中哪一步是控制步骤,是受传质影响还是受传荷影响;在氯化物或硫酸盐体系中,三价铬的还原历程是否一致,又或者三价铬的还原历程受配位剂、pH 的影响更大。以上都是值得深入研究的问题。
(3) 加强离子液体中三价铬电沉积的研究。三价铬在水溶液中电沉积功能铬的问题较多,困难较大。离子液体具有较宽的电化学窗口,电沉积过程中副反应少,引起了人们的广泛关注。近年来有资料报道[40],在离子液体中电沉积三价铬可得到厚镀层,阴极电流效率30%~40%,当电流密度为15 A/dm2时,沉积速率0.8~1.0 µm/min。在离子液体中电沉积三价铬将成为研究热点。
[1]王秋红,潘湛昌,胡光辉,等.三价铬镀液电镀铬的工艺研究[J].电镀与精饰,2010,32 (11):13-16.
[2]杨哲龙,屠振密,张景双,等.三价铬电镀的新进展[J].电镀与环保,2001,21 (2):1-4.
[3]屠振密,郑剑,李宁,等.三价铬电镀铬现状及发展趋势[J].表面技术,2007,36 (5):59-63,87.
[4]SURVILIENE S,JASULAITIENE V,NIVINSKIENE O,et al.Effect of hydrazine and hydroxylaminophosphate on chrome plating from trivalent electrolytes [J].Applied Surface Science,2007,253 (16):6738-6743.
[5]SZIRÁKI L,KUZMANN E,PAPP K,et al.Electrochemical behaviour of amorphous electrodeposited chromium coatings [J].Materials Chemistry and Physics,2012,133 (2/3):1092-1100.
[6]杜登学,张长鑫.二甲基甲酰胺体系三价铬镀铬的研究[J].材料保护,1995,28 (5):19-21.
[7]王华,曾振欧,赵国鹏,等.硫酸盐溶液体系中三价铬镀厚铬工艺及镀层性能研究[J].电镀与涂饰,2007,26 (6):13-17.
[8]侯峰岩,屠振密,屈云腾.环保型低浓度硫酸盐三价铬电沉积厚铬的研究[J].复旦学报(自然科学版),2012,51 (2):168-171.
[9]SURVILIENE S,LISOWSKA-OLEKSIAK A,SELSKIS A,et al.Corrosion behavior of Cr coatings deposited from Cr(III) formate-urea electrolytes [J].Transactions of the Institute of Metal Finishing,2006,84 (5):241-245.
[10]KUZNETSOV V V,VINOKUROV E G,KUDRYAVTSEV V N.Effect of hydrodynamic electrolysis conditions on the kinetics of cathodic processes in chromium(III) sulfate electrolytes [J].Russian Journal of Electrochemistry,2000,36 (7):756-760.
[11]曾振欧,康振华,赵国鹏.三价铬硫酸盐溶液厚铬镀层性能研究[J].电镀与涂饰,2010,29 (6):8-11.
[12]DANILOV F I,PROTSENKO V S,GORDIIENKO V O,et al.Nanocrystalline hard chromium electrodeposition from trivalent chromium bath containing carbamide and formic acid:Structure,composition,electrochemical corrosion behavior,hardness and wear characteristics of deposits [J].Applied Surface Science,2011,257 (18):8048-8053.
[13]TU Z M,YANG Z L,ZHANG J S,et al.Cathode polarization in trivalent chromium plating [J].Plating and Surface Finishing,1993,79 (9):78-82.
[14]屠振密,杨哲龙,汪沧海,等.三价铬电镀机理的研究──铬层不能增厚的原因[J].材料保护,1985,18 (3):14-16,8.
[15]屠振密,杨哲龙,汪沧海,等.三价铬镀液电解时阴极附近pH 值的测定[J].电镀与环保,1984 (5):21-25.
[16]SURVILIENĖ S,NIVINSKIENĖ O,ČEŠUNIENĖ A,et al.Effect of Cr(III) solution chemistry on electrodeposition of chromium [J].Journal of Applied Electrochemistry,2006,36 (6):649-654.
[17]艾仕云.三价铬电镀厚铬镀层的研究[J].电镀与环保,1997,17 (3):5-7.
[18]李国华,赖奂汶,黄清安.三价铬镀液中配体的作用[J].材料保护,2005,38 (12):44-46.
[19]BARAL A,ENGELKEN R.Modeling,optimization,and comparative analysis of trivalent chromium electrodeposition from aqueous glycine and formic acid baths [J].Journal of the Electrochemical Society,2005,152 (7):C504-C512.
[20]SAFONOV V A,VYKHODTSEVA L N,POLUKAROV Y M,et al.Valence-to-core X-ray emission spectroscopy identification of carbide compounds in nanocrystalline Cr coatings deposited from Cr(III) electrolytes containing organic substances [J].Journal of Physical Chemistry B,2006,110 (46):23192-23196.
[21]SZYNKARCZUK J,DRELA I,KUBICKI J.Electrochemical behaviour of chromium(III) in the presence of formic acid—I [J].Electrochimica Acta,1989,34 (3/4):399-403.
[22]ZENG Z X,SUN Y L,ZHANG J Y.The electrochemical reduction mechanism of trivalent chromium in the presence of formic acid [J].Electrochemistry Communications,2009,11 (2):331-334.
[23]GIOVANARDI R,ORLANDO G.Chromium electrodeposition from Cr(III) aqueous solutions [J].Surface and Coatings Technology,2011,205 (15):3947-3955.
[24]EL-SHARIF M,MA S,CHISHOLM C U.Environmentally acceptable process for electrodeposition of hard chromium from chromium(III) electrolyte [J].Transactions of the Institute of Metal Finishing,1995,73 (1):19-25.
[25]EL-SHARIF M.Replacing hexavalent chromium in electroplating [J].Transactions of the Institute of Metal Finishing,1997,75 (6):B143-B146.
[26]IBRAHIM S K,WATSON A,GAWNE D T.The role of formic acid and methanol on speciation rate and quality in the electrodeposition of chromium from trivalent electrolytes [J].Transactions of the Institute of Metal Finishing,1997,75 (5):181-188.
[27]ZENG Z X,ZHANG Y X,ZHAO W J,et al.Role of complexing ligands in trivalent chromium electrodeposition [J].Surface and Coatings Technology,2011,205 (20):4771-4775.
[28]HONG G,SIOW K S,ZHIQIANG G,et al.Hard chromium plating from trivalent chromium solution [J].Plating and Surface Finishing,2001,88 (3):69-75.
[29]ZENG Z X,WANG L P,LIANG A M,et al.Tribological and electrochemical behavior of thick Cr-C alloy coatings electrodeposited in trivalent chromium bath as an alternative to conventional Cr coatings [J].Electrochimica Acta,2006,52 (3):1366-1373.
[30]HUANG C A,LIU Y W,CHUANG C H.The hardening mechanism of a chromium-carbon deposit electroplated from a trivalent chromium-based bath [J].Thin Solid Films,2009,517 (17):4902-4904.
[31]GHAZIOF S,GOLOZAR M A,RAEISSI K.Characterization of as-deposited and annealed Cr-C alloy coatings produced from a trivalent chromium bath [J].Journal of Alloys and Compounds,2010,496 (1/2):164-168.
[32]KWON S C,KIM M,PARK S U,et al.Characterization of intermediate Cr-C layer fabricated by electrodeposition in hexavalent and trivalent chromium baths [J].Surface and Coatings Technology,2004,183 (2/3):151-156.
[33]PROTSENKO V S,GORDIIENKO V O,DANILOV F I.Unusual “chemical” mechanism of carbon co-deposition in Cr-C alloy electrodeposition process from trivalent chromium bath [J].Electrochemistry Communications,2012,17:85-87.
[34]PROTSENKO V S,DANILOV F I,GORDIIENKO V O,et al.Electrodeposition of hard nanocrystalline chrome from aqueous sulfate trivalent chromium bath [J].Thin Solid Films,2011,520 (1):380-383.
[35]DUAN S,LI H,ZHANG X,et al.Hard chromium plating from a trivalent plating bath [J].Plating and Surface Finishing,1995,82 (6):84-86.
[36]李惠东,段淑贞,张新.三价铬镀液电镀硬铬研究[J].电镀与涂饰,1993,12 (4):5-8.
[37]ZENG Z X,LIANG A M,ZHANG J Y.A review of recent patents on trivalent chromium plating [J].Recent Patents on Materials Science,2009,2 (1):50-57.
[38]PROTSENKO,GORDIIENKO V O,DANILOV F I,et al.Thick chromium electrodeposition from trivalent chromium bath containing carbamide and formic acid:An investigation into current efficiency,electrodeposition rate and surface morphology [J].Metal Finishing,2011,109 (4/5):33-37.
[39]曾志翔,梁爱民,张俊彦.三价铬电镀硬铬工艺的中试研究[J].电镀与环保,2008,28 (2):17-20.
[40]BENABEN P.An overview of hard chromium plating using trivalent chromium solutions [R/OL][2011-01-31].http://www.pfonline.com/ articles/an-overview-of-hard-chromium-plating-using-trivalent-chromiumsolutions.
[41]CHOI Y,KIM M,KWON S C.Characterization of chrome layer formed by pulse plating [J].Surface and Coatings Technology,2003,169/170:81-84.
[42]SARAVANAN G,MOHAN S.Corrosion behavior of Cr electrodeposited from Cr(VI) and Cr(III)-baths using direct (DCD) and pulse electrodeposition (PED) techniques [J].Corrosion Science,2009,51 (1):197-202.
[43]HE X K,QIU G Z,CHEN B Z,et al.Process of pulse electrodeposition nanocrystalline chromium from trivalent chromium bath [J].Transactions of Nonferrous Metals Society of China,2007,17:s685-s691.
[44]SONG Y B,CHIN D T.Pulse plating of hard chromium from trivalent baths [J].Plating and Surface Finishing,2000,87 (9):80-87.
[45]FENG H,ZHANG Y,WEI Y L,et al.Study of single-metal multi-nanometer electrodeposition chromium layer [J].International Journal of Hydrogen Energy,2009,34 (2):1114-1118.
猜你喜欢
湿法冶金(2020年1期)2020-02-24
科学与财富(2017年9期)2017-06-09
中国氯碱(2017年3期)2017-04-18
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
电镀与环保(2016年2期)2017-01-20
物理化学学报(2015年7期)2015-12-30
表面工程与再制造(2014年2期)2014-02-27
表面工程与再制造(2014年2期)2014-02-27
表面工程与再制造(2014年2期)2014-02-27
