
水稻Trihelix转录因子家族全基因组分析及功能预测
2015-10-29 07:01纪剑辉周颖君吴贺贺杨立明
遗传 2015年12期
纪剑辉,周颖君,吴贺贺,杨立明
水稻Trihelix转录因子家族全基因组分析及功能预测
纪剑辉,周颖君,吴贺贺,杨立明
淮阴师范学院生命科学学院,江苏省环洪泽湖生态农业生物技术重点实验室,江苏省区域现代农业与环境保护协同创新中心,淮安 223300
Trihelix转录因子家族在植物生长发育以及响应逆境胁迫等方面发挥着重要作用,但目前基于水稻全基因组水平鉴定和分析该基因家族的研究尚未见相关报道。本文利用生物信息学方法在水稻基因组数据库中鉴定到Trihelix家族成员31个,序列聚类和功能结构域分析发现该家族均含有高度保守的、特征性的Trihelix结构域;根据亲缘关系远近和结构域特点,将其分为5个亚家族(Ⅰ~Ⅴ)。通过与拟南芥、二穗短炳草和高粱中Trihelix家族的聚类分析发现,这4个物种中Trihelix家族的分类相一致,但每个物种均含有不同亚家族的成员,表明该基因家族的分化早于物种的分化。基于MEME程序分析水稻Trihelix转录因子家族的保守基序与聚类分析结果具有较高的一致性。染色体区段复制分析表明,部分Trihelix家族成员在水稻以及水稻与其他物种之间存在种内和种间的染色体区段复制;生物芯片数据分析发现,Trihelix基因家族在水稻不同组织中、以及对6种不同植物激素的响应呈现多样化的表达谱。采用RiceFREND在线数据库分析发现,水稻Trihelix转录因子家族的20个成员与其他蛋白存在互作关系。本研究结果初步明确了水稻Trihelix转录因子家族的进化特点、染色体分布、染色体区段复制关系、组织表达、激素应答,以及该家族蛋白与其他蛋白质的互作情况,为进一步揭示Trihelix转录因子家族的分子进化规律和生物学功能奠定了基础。
水稻;Trihelix;转录因子家族;进化
转录因子(又称反式作用因子)是一类DNA结合蛋白,能够与靶标基因启动子区域的顺式作用元件发生特异性相互作用,从而调控靶标基因的表达。目前在植物中已发现60多个转录因子家族,其中Trihelix是植物中最早发现的转录因子家族之一,因其保守结构域含有3个串联的α螺旋而得名[1,2]。Kaplan-Levy等[3]根据Trihelix家族的保守域结构特征将Trihelix家族分为5个亚家族,分别为GT-1、GT-2、SH4、GTγ和SIP1,每个亚家族的名称根据该亚家族第一个被发现的成员名字来命名。已有研究报道Trihelix转录因子在植物生长发育过程和响应逆境胁迫中起着重要调控作用[4~7]。豌豆(L)GT-1因子是最早被鉴定的Trihelix转录因子,能与光诱导基因启动子上的GT元件特异性结合[4],进而调控基因的表达,因此也被称为GT因子家族[4]。随后在烟草(L.)[5]、拟南芥()[6]和水稻()[7]中均克隆出该基因的同源基因。此外,在水稻中鉴定出一个类似于GT-1因子的DNA结合蛋白GT-2,与水稻光敏色素A基因对光处理的应答相关[7],相较于GT-1因子,该蛋白含有2个重复的保守结构域[8,9]。拟南芥的()属于GT-2家族,该基因能够调控拟南芥花瓣和萼片的生长,这也是Trihelix家族中发现的首个与花器官形态相关基因[10~12]。水稻()基因是目前鉴定出的唯一一个SH4亚家族成员,该基因编码一个SH4类型的转录因子,在细胞分化激活中扮演重要作用,该基因的突变体在栽培水稻中导致种子落粒性的消失[13]。GTγ亚家族中目前只鉴定出4个水稻GTγ成员,分别是、和,其中大部分基因已被证实与冷、干旱和盐胁迫相关[14]。SIP1亚家族的部分成员已在烟草、拟南芥中被克隆和鉴定,与植物的胚胎发育、叶片发育以及细胞增殖相关[15~17]。
为进一步了解Trihelix转录因子家族的分子特征、基因组分布及其可能的生物学功能,本文利用生物信息学方法鉴定和分析了水稻Trihelix转录因子家族,并从基因组水平上对水稻Trihelix基因家族的序列特征、进化规律、染色体定位和染色体区段复制、组织表达、激素应答以及蛋白互作等方面进行了系统地分析与预测,从而为深入分析水稻Trihelix转录因子基因家族的进化规律和生物学功能提供依据。
1 材料和方法
1.1 Trihelix基因家族数据获取与分析
从水稻TIGR基因组数据库下载水稻全基因组序列,以模式植物拟南芥Trihelix蛋白在Pfam数据库(http://pfam.sanger. ac.uk/)中检索Trihelix基因家族蛋白特征结构域[18](标号PF13837),利用HMMER3程序(http://hmmer.janelia.org/)以PF13837隐马可夫模型在水稻全基因组序列中搜索含有Trihelix结构域特征的序列(E≤10-10被认为是候选蛋白)。利用SMART在线程序检测候选蛋白,去除不含Trihelix结构域特征的序列[19],即获得水稻Trihelix转录因子家族序列,同时利用ExPASy数据库(http:// www.expasy.org/)对水稻Trihelix蛋白的分子量、等电点等基本信息进行分析[20]。基于上述方法在TFDB数据库(http://planttfdb.cbi.edu.cn/index.php, v3.0)[21]获得拟南芥、二穗短柄草()和高粱()的Trihelix基因序列。
在RGAP数据库(http://rice.plantbiology.msu.edu/)获取上述水稻Trihelix蛋白基因的CDS序列,并在KOME数据库(http://cdna01.dna.affrc.go.jp/cDNA/)中获得对应的Trihelix基因序列号。
1.2 Trihelix保守基序的鉴定和分析
利用蛋白质保守基序在线搜索程序MEME (http://meme.nbcr.net/meme/cgi-bin/meme.cgi)分析水稻Trihelix转录因子家族的保守基序,并设置相关参数为基序重复的数量为“any”,基序长度为6~200 aa,预测基序的数量为25个[22,23]。
1.3 多序列联配、蛋白质保守序列比对和系统进化树的构建
利用Cluster 3.0程序对候选水稻Trihelix转录因子家族保守区域氨基酸进行多序列联配分析[24],序列比对结果通过Mega5程序[25](http://www.mega-software.net/)的邻接法(Neighbor-joining method, NJ)构建水稻Trihelix转录因子保守区域的系统进化树,Bootstrap值设为1000。利用WebLogo 3.3程序对水稻Trihelix转录因子保守区域氨基酸进行分析[26]。采用邻接法构建水稻、拟南芥、二穗短柄草和高粱中Trihelix转录因子家族的系统进化树。
1.4 Trihelix基因家族在不同物种中染色体区段的复制分析
利用植物基因组复制数据库PGDD(http://chibba. agtec.uga.edu/duplication/)[27]分析候选水稻Trihelix基因所在染色体区段的复制情况,结果通过基因染色体定位软件MapInspect (http://www.dpw.wau.nl/pv/ pub/MapComp/)进行图示化;利用相同方法分析水稻与二穗短柄草、水稻与高粱之间Trihelix基因所在染色体区段的复制情况,所得结果通过Circos软件 (http://circos.ca/)进行图示化[28]。
1.5 水稻Trihelix基因家族的不同组织表达以及对不同植物激素的响应分析
利用水稻全基因芯片数据[29](http://ricexpro.dna. affrc.go.jp/data-set.html)以及表达谱芯片分析系统(Agilent one-color (Cy3) microarray-based gene analysis system)获得水稻Trihelix基因家族在叶片、叶鞘、根、茎、穗、花药、雌蕊、外稃、内稃、胚珠、胚和胚乳等不同组织中的表达结果,每组包含3个重复。利用水稻RiceXPRO芯片数据获取水稻根组织在脱落酸(Abscisic acid)、赤霉素(Gibberellin)、生长素(Auxin)、油菜素甾醇(Brassinosteroid)、细胞分裂素(Cytokinin)和茉莉酸(Jasmonic acid ) 6种植物激素处理下水稻Trihelix基因家族的表达结果。水稻Trihelix基因家族的不同组织表达以及对不同植物激素的响应结果均采用RiceXPro内置Cluster 3.0程序进行层次聚类,并由RiceXPro提供的R程序中的"gplots"程序包进行Heatmap图示。
1.6 水稻Trihelix转录因子的蛋白互作网络
利用水稻RiceFREND在线数据库(http://ricefrend. dna.affrc.go.jp/)分析水稻Trihelix转录因子与其他蛋白的互作情况,结果采用Graphviz软件(Graph Visualization Software)作图[30]。
2 结果与分析
2.1 水稻Trihelix转录因子家族的鉴定
在水稻基因组数据库中共鉴定出31个Trihelix转录因子(表1),其中最长的序列有882个氨基酸残基,最短序列有89个氨基酸残基,等电点范围为4.1264~10.7441。WebLogo图示化结果显示,31个Trihelix转录因子均含有典型的α螺旋结构,并且氨基酸残基Trp(W)-1、Trp(W)-69和Cys(C)-102在这些序列中高度保守(图1)。
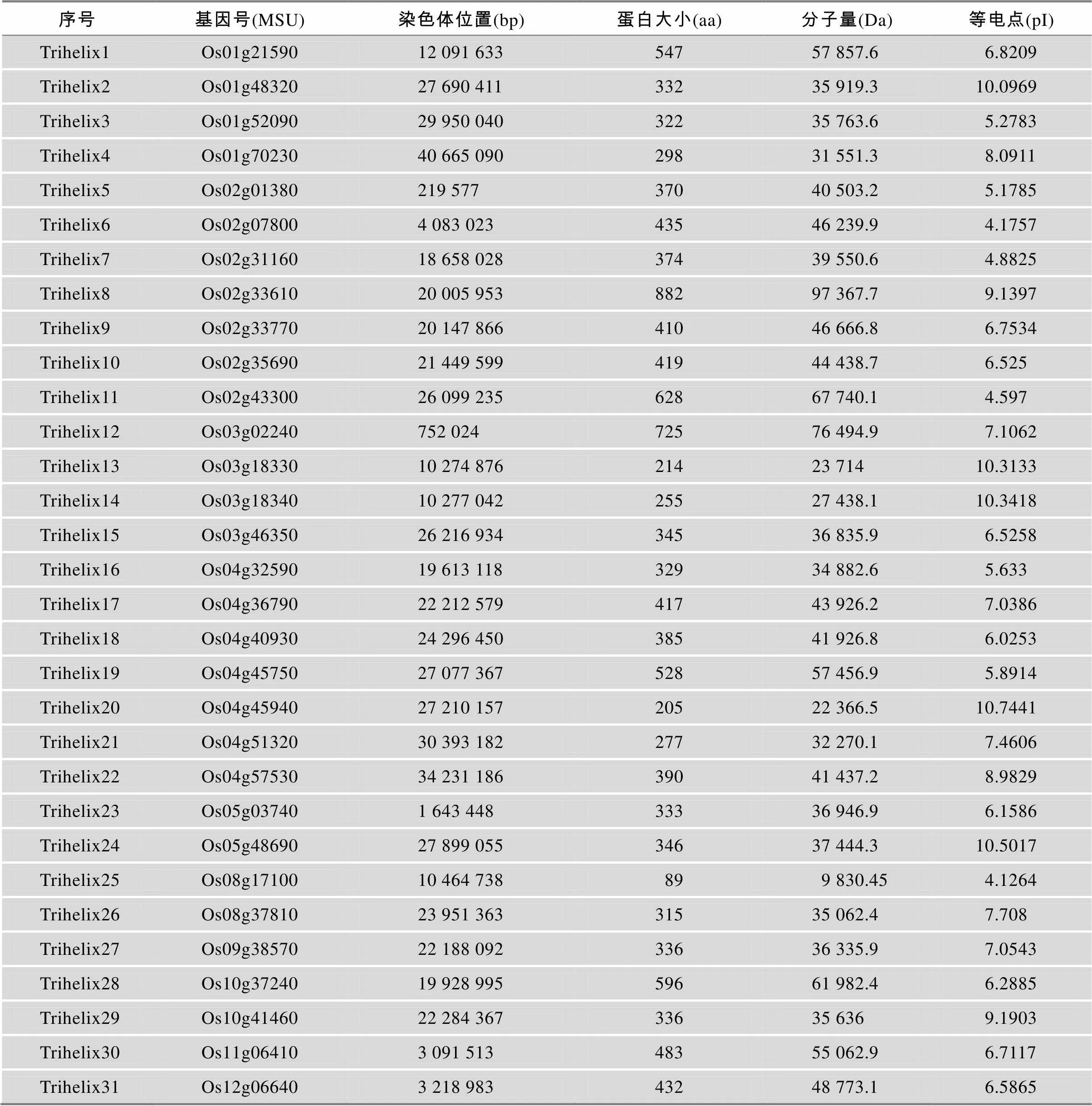
表1 水稻Trihelix转录因子家族基因基本信息
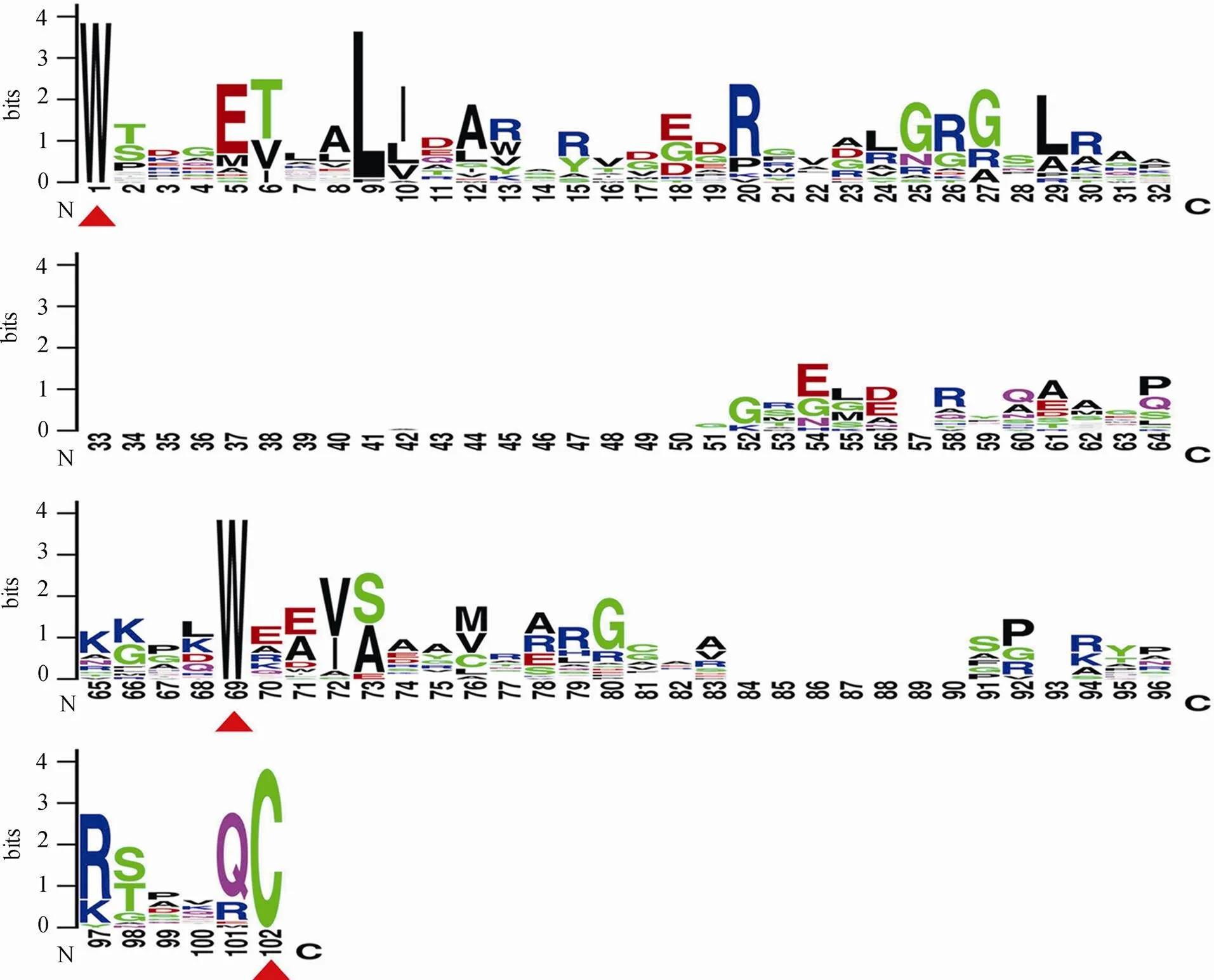
图1 Trihelix家族特征结构域在水稻中的保守性分析
序列中不同位点氨基酸的堆叠高度表明该位点的保守程度,堆叠中单一氨基酸的高度显示该种氨基酸在该位置的相对频率。红色三角显示保守核心氨基酸Trp(W)-1、Trp(W)-69和Cys(C)-102。
2.2 Trihelix转录因子家族的进化分析
为了解水稻Trihelix转录因子家族成员的进化关系,以水稻Trihelix蛋白的保守域序列构建了系统进化树。结果显示水稻Trihelix转录因子家族分成5个亚家族(Ⅰ~Ⅴ)(图2),其中亚家族Ⅰ、Ⅱ在亲缘关系上更为相近。同样,拟南芥、二穗短炳草和高粱等3个物种的Trihelix转录因子家族也分成5个亚家族(Ⅰ~Ⅴ)(图3)。除Trihelix25外,水稻和其他植物中的Trihelix家族基因并非单独聚类并独立进化,而是每个亚家族都同时含有水稻和其他植物中的Trihelix成员,并且每个亚家族的成员具有相似的结构域。由此表明在水稻、高粱、拟南芥和二穗短柄草等物种分化之前,这些Trihelix基因已在同一物种中发生了分化。此外,从每个亚家族的基因分布量上看,Ⅳ亚家族的基因数量远多于其他亚家族基因数量。
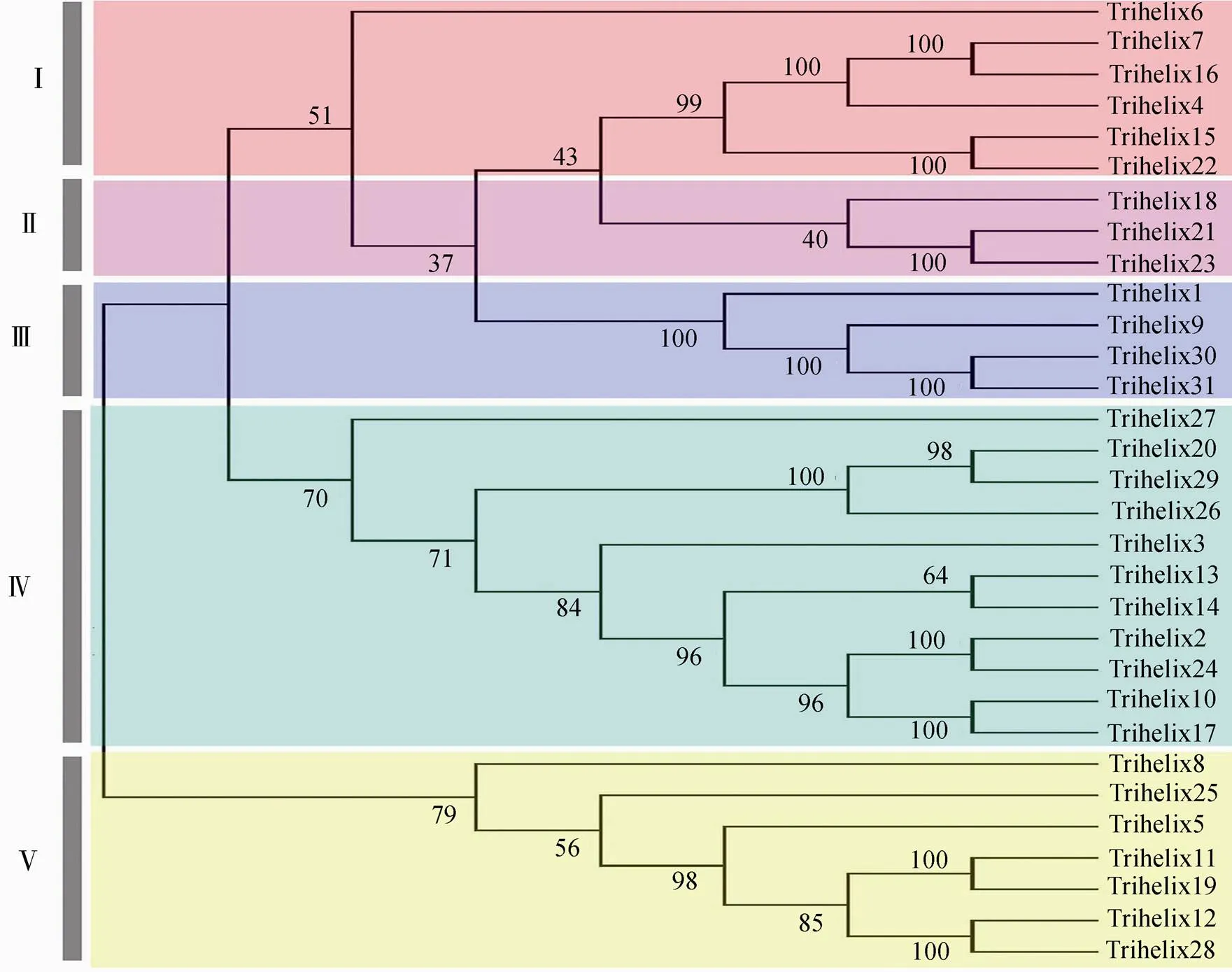
图2 水稻Trihelix转录因子家族的进化树分析
系统进化树采用邻接法由MEGA 5.0程序构建,Bootstrap值设置为1000。
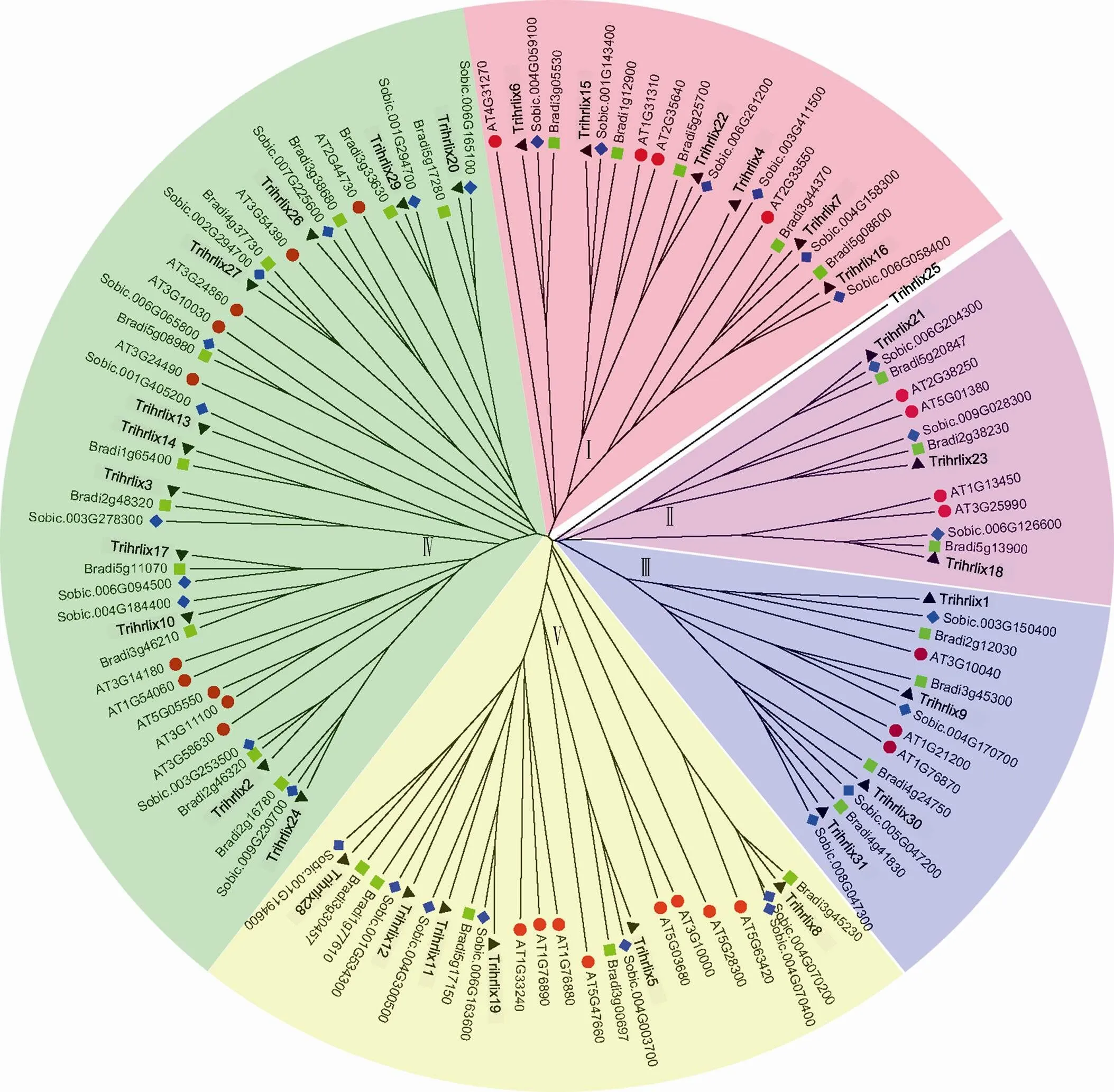
图3 水稻、拟南芥、二穗短柄草和高粱中Trihelix转录因子家族进化分析
黑色三角代表水稻Trihelix家族基因;红色圆形代表拟南芥Trihelix家族基因;绿色方形代表二穗短柄草Trihelix家族基因,蓝色棱形代表高粱Trihelix家族基因。
水稻Trihelix转录因子家族的基序分析结果显示,水稻Trihelix转录因子含有Trihelix家族典型的保守基序Motif1和Motif2(Trihelix25不含有Motif1)。此外,还有其他一些相对保守的基序,这些基序最少含6个氨基酸残基,最多含79个氨基酸残基(表2)。
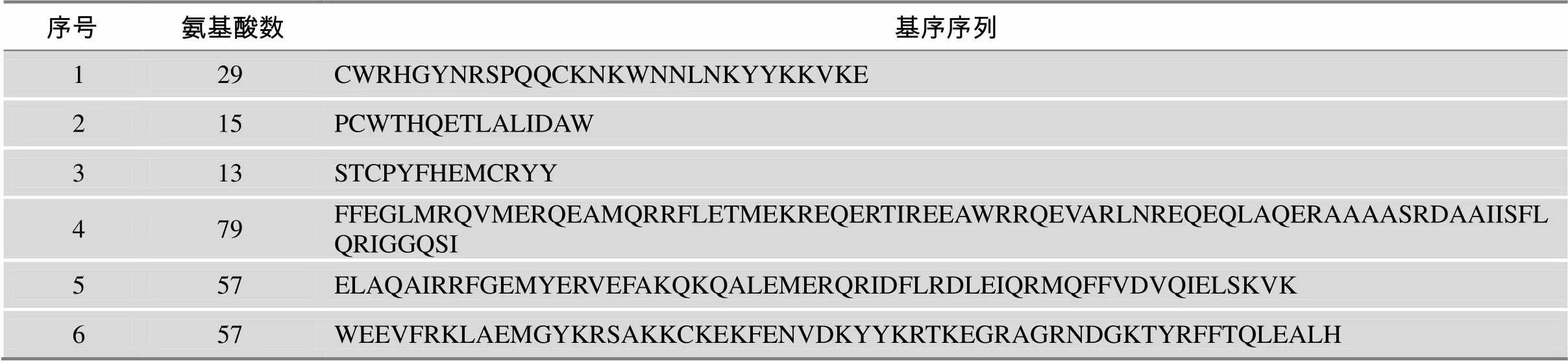
表2 水稻Trihelix转录因子的保守基序
水稻Trihelix转录因子的基序特征与进化树基本保持一致。从图4可以看出,亚家族Ⅰ中所有蛋白均含有Motif7基序,亲缘关系更为接近的Trihelix4、7和16的C端含有Motif16基序。亚家族Ⅲ中Trihelix1、9、30、31都含有Motif12基序,且均紧邻Motif1基序;亲缘关系更为接近的Trihelix9、30、31均含Motif21基序。此外,Trihelix30相较于Trihelix31仅在其N末端多出Motif25基序,其余完全一致。从亚家族Ⅳ中亲缘关系较近的Trihelix10、17可以看出,Trihelix17比Trihelix10多出3个Motif10基序,其他部分基本一致;从亚家族Ⅴ中亲缘关系较近的Trihelix11、19、12、28中也可以看出这个几个转录因子的C端基序整体结构相似性较高。
2.4 水稻Trihelix转录因子家族所在染色体区段复制分析
染色体区段复制分析发现,水稻所有染色体均分布有Trihelix基因,其中2号和4号染色体各分布7个基因,9、11和12号染色体上各分布1个基因(图5)。此外,水稻31个Trihelix基因在染色体上的分布存在聚集现象,如在2号染色体的20 Mb、3号染色体的10 Mb、4号染色体的27 Mb区段位置均存在2个或2个以上基因聚集。总体而言,发生复制的7组水稻Trihelix基因均属于同一个亚家族,并且在进化树的位置均较为靠近,显示其具有较高的同源性(表3)。
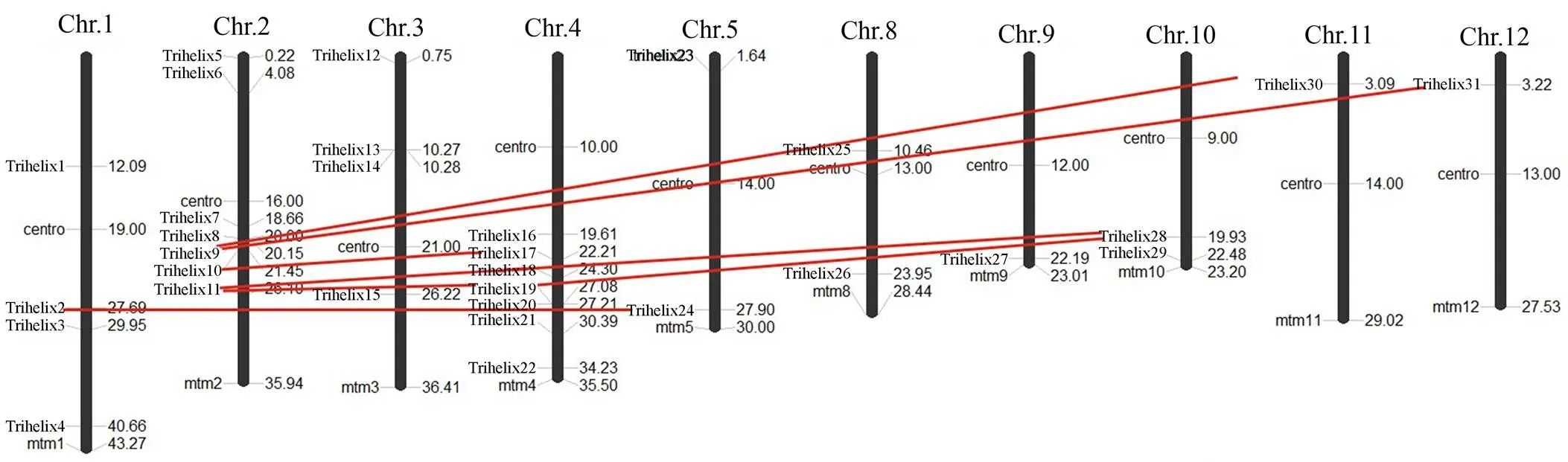
图5 水稻Trihelix转录因子家族在染色体上的复制分析
染色体的物理位置(Mb)在每一条染色体的右侧显示;水稻中所有的Trihelix基因则被标注在染色体的左侧,基因组进化过程中同源复制事件用直线连接表示;整条染色体的长度用mtm(1~12)来标注,复制的Trihelix基因用红色连线来显示。
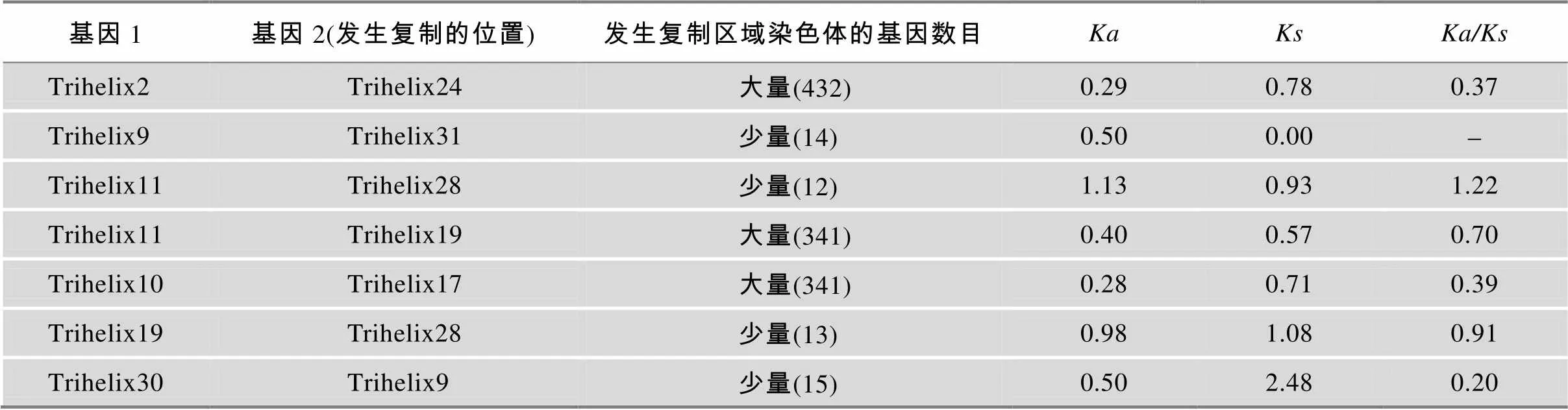
表3 水稻Trihelix转录因子家族在染色体上的复制分析
2.5 Trihelix转录因子家族在水稻和高粱、水稻和二穗短柄草之间染色体区段复制分析
Trihelix转录因子家族在水稻和高粱、水稻和二穗短柄草之间的染色体复制分析结果显示,水稻中有28个Trihelix基因(Trihelix2、22和25除外)与二穗短柄草中的Trihelix基因具有线性同源关系(图6A),有28个Trihelix基因(Trihelix8、14和25除外)与高粱中的Trihelix基因具有线性同源关系(图6B)。推测这些基因在进化过程中经历了复制、重排和缺失等一系列进化事件。
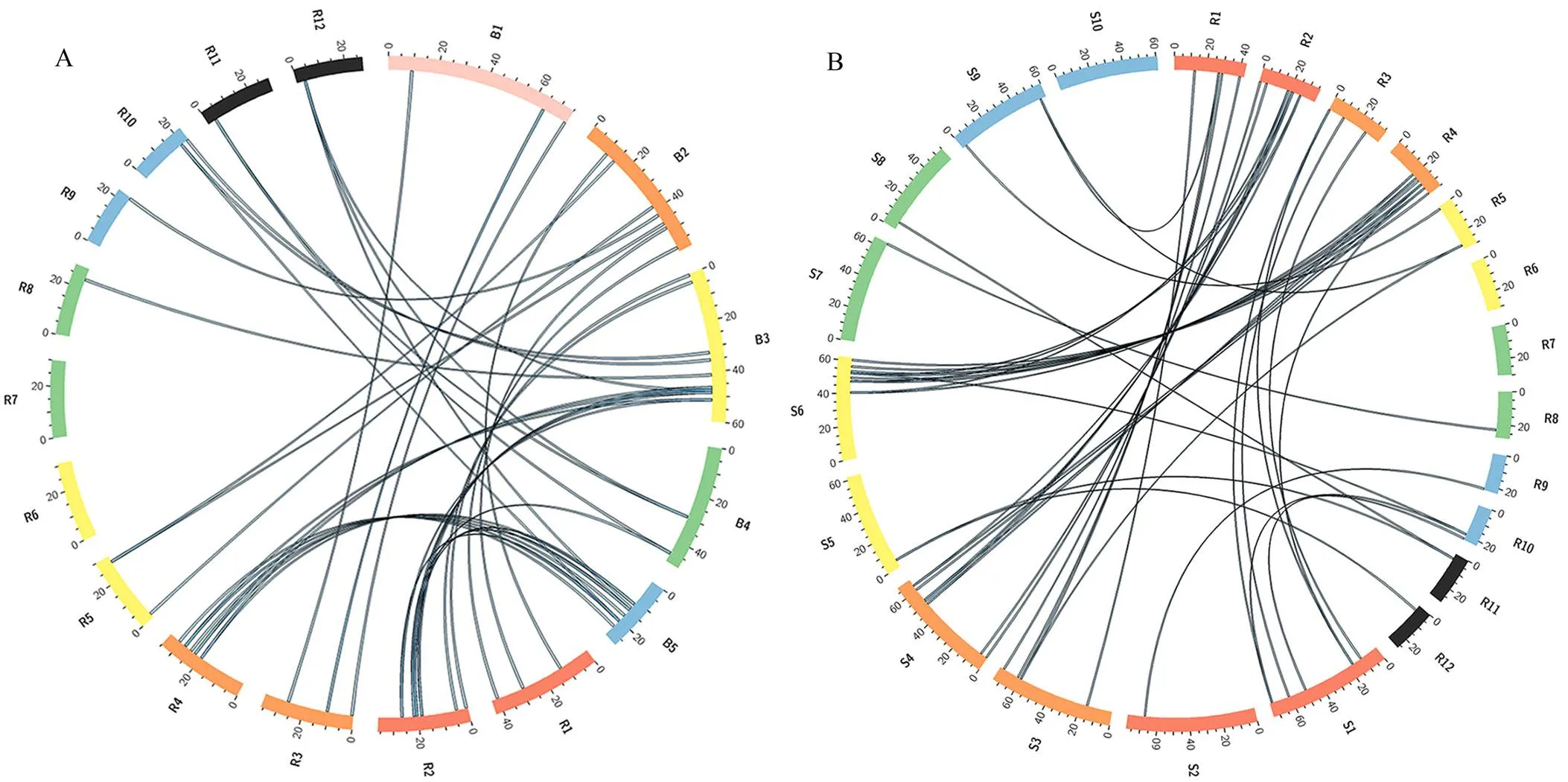
图6 Trihelix基因在水稻和二穗短柄草、水稻和高粱中的染色体线性复制分析
A:Trihelix基因在水稻和二穗短柄草基因组中的染色体线性复制分析 (水稻染色体用R1~R12来表示,二穗短柄草染色体用B1~B5来表示);B:Trihelix基因在水稻和高粱基因组中的染色体线性复制分析 (水稻染色体用R1~R12来表示,高粱染色体用S1~S10来表示)。发生复制的Trihelix基因则用黑色连线来显示。
2.6 Trihelix转录因子家族在水稻不同组织中的表达模式分析
Trihelix转录因子家族在水稻不同组织的表达模式分析结果显示,相关基因的表达几乎涉及水稻不同组织的整个生长过程,27个Trihelix基因(Trihelix4、15、22和25无芯片数据)根据其表达情况经聚类分析后分成7个不同的亚类,不同的亚类具有相似的表达谱变化。从表达谱的分析结果可以看出,第Ⅶ亚类的Trihelix11、12、19和28在叶片、根的部分时期和花序中的表达量较低,在外稃和内稃、子房早期和胚乳后期中表达相对较高,提示第Ⅶ亚类可能更多和生殖生长有关;第Ⅵ亚类的Trihelix5和24在花序、花药、雌蕊、外稃和内稃中表达较高,在其他组织中表达相对较低(图7),但在聚类分析中Trihelix5和24则分别位于Ⅴ和第Ⅳ亚家族(图2);第Ⅴ亚类的Trihelix1、2、9、20和30在叶片、叶鞘和茎中表达相对较低,在子房、胚和胚乳中表达相对较高,其中Trihelix20为第Ⅲ亚家族成员,Trihelix1、9和30则均为第Ⅱ亚家族成员;除以上几个亚类,其他亚类的基因表达则相对分散。
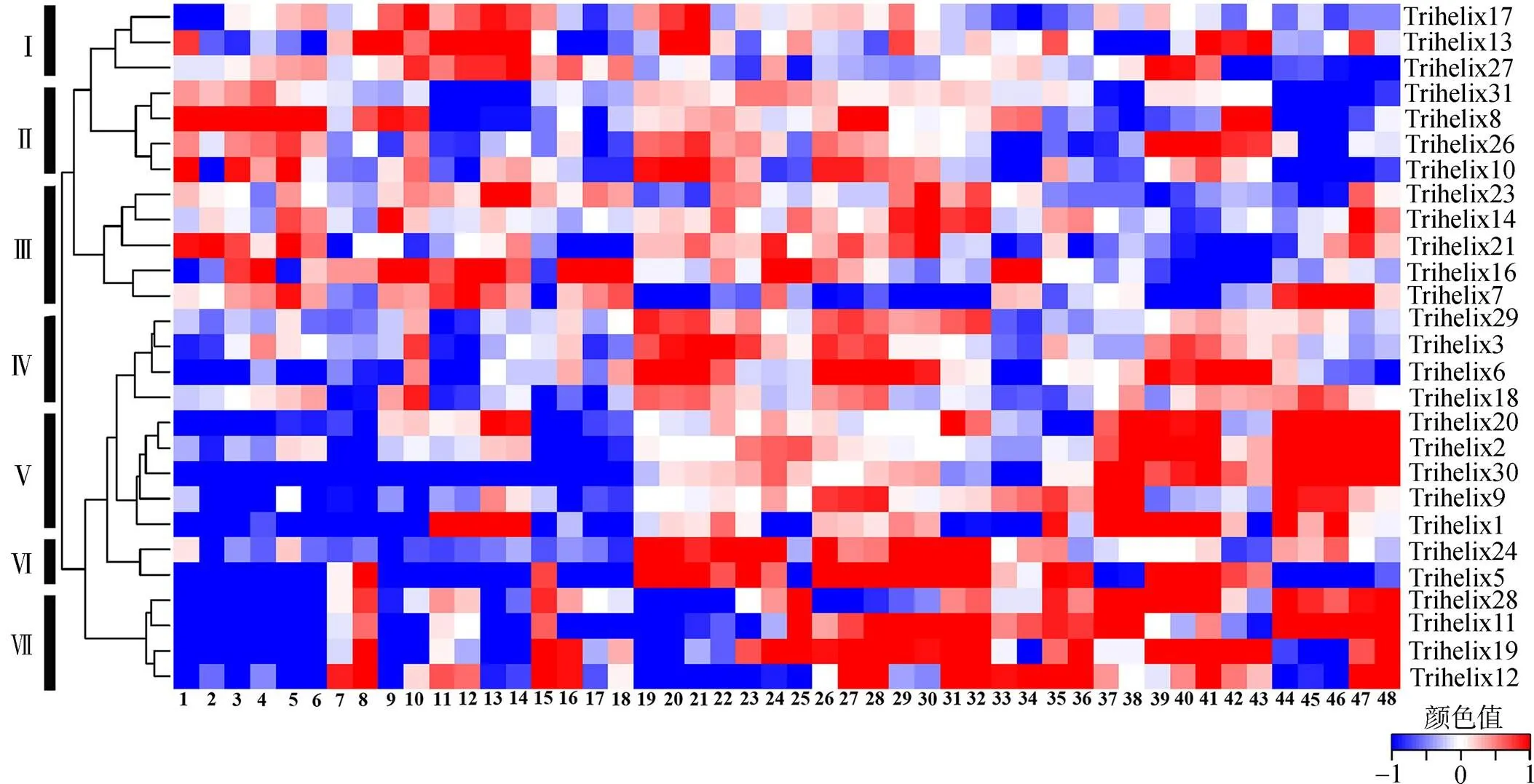
图7 水稻Trihelix转录因子家族在不同组织及不同生长阶段的表达聚类分析
1:27 d生长叶片;2:28 d生长叶片;3: 76 d生长叶片;4: 77 d生长叶片;5: 125d旗叶;6: 126 d旗叶;7: 27 d叶鞘;8: 28 d叶鞘;9: 76 d叶鞘;10: 77 d叶鞘;11: 27 d根部;12: 28 d根部;13: 76 d根部;14: 77 d根部;15: 26 d茎部组织;16: 27 d茎部组织;17: 76 d茎部组织;18: 77 d茎部组织;19:花序(0.6~1.0 mm);20: 花序(3.0~4.0 mm);21: 花序(5.0~10.0 mm);22: 花药(0.3~0.6 mm);23: 花药(0.7~1.0 mm);24: 花药(1.2~1.5 mm);25: 花药(1.6~2.0 mm);26: 雌蕊(穗长5~10cm);27: 雌蕊(穗长10~14 cm);28: 雌蕊(穗长14~18 cm);29: 外稃(小花1.5~2.0 mm);30: 内稃(小花1.5~2.0 mm);31: 外稃(小花长4.5-5.0 mm);32: 内稃(小花长4.5~5.0 mm);33: 外稃(小花长7.0 mm);34: 内稃(小花长7.0 mm);35: 子房(开花后1 d);36: 子房(开花后3 d);37: 子房(开花后5 d);38: 子房(开花后7 d);39: 胚(开花后7 d);40: 胚(开花后10 d);41: 胚(开花后14 d);42: 胚(开花后28 d);43: 胚(开花后42 d);44: 胚乳(开花后7 d);45: 胚乳(开花后10 d);46: 胚乳(开花后14 d);47: 胚乳(开花后28 d);48:胚乳(开花后42 d);彩色棒状图中的颜色显示基因表达强度, 红色代表最高, 白色代表居中,蓝色代表最低。
2.7 6种不同激素处理后Trihelix基因在水稻根组织中的表达分析
生物芯片的数据分析显示,水稻根组织在经过脱落酸处理后Trihelix2、5、11、13、19和21的表达有明显的增加,Trihelix1和12则呈明显的减少;经生长素处理后Trihelix11和12呈明显减少;经油菜素甾醇处理之后仅Trihelix1的表达量呈明显减少;经细胞分裂素处理后Trihelix1和28呈明显减少,Trihelix11则呈明显增加;在经茉莉酸处理后Trihelix9、20、21和27呈明显增加,除此之外,其他基因没有明显的变化(图8)。
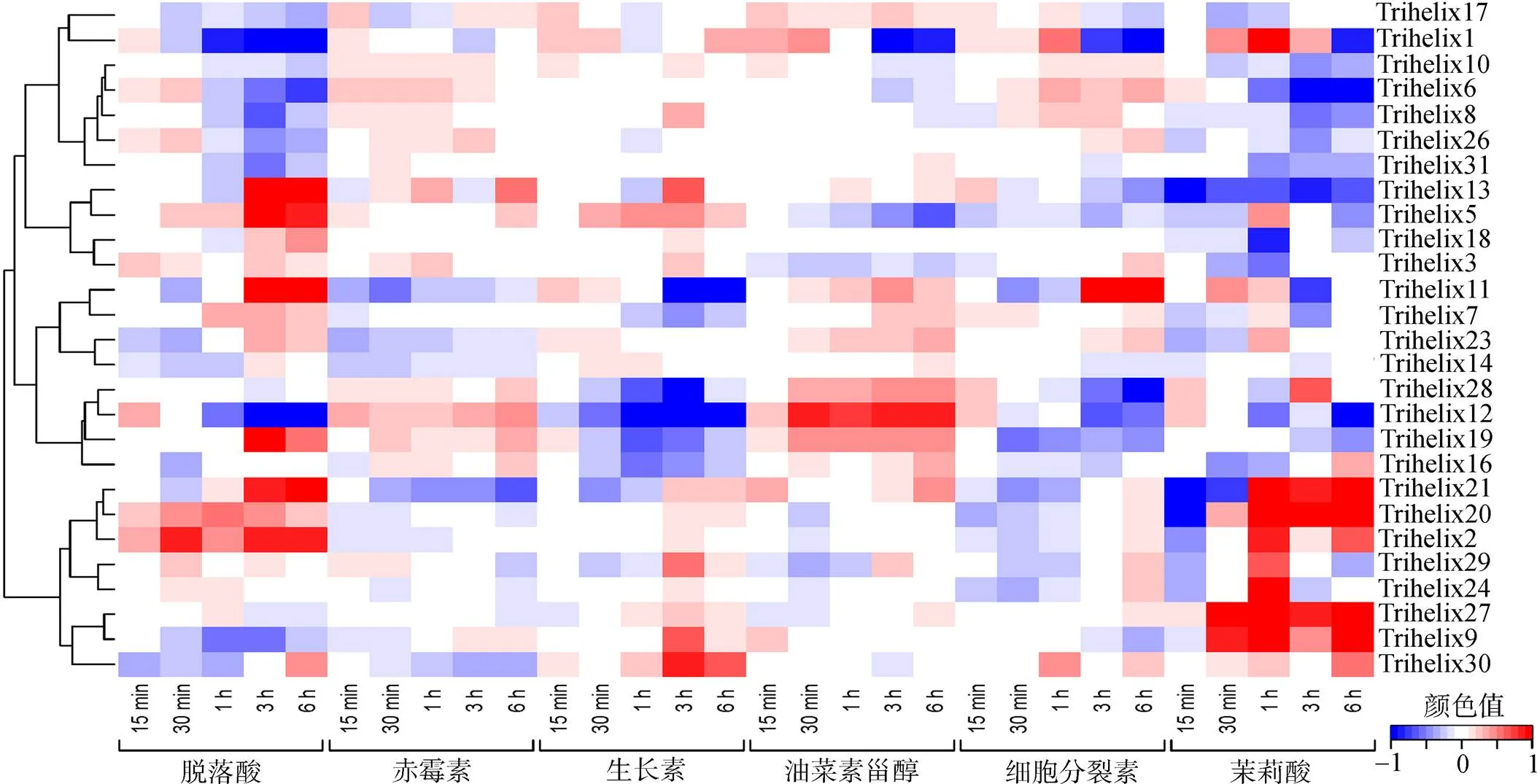
图8 6种植物激素处理后水稻根部组织中Trihelix基因表达分析
彩色棒状图中的颜色显示基因表达强度,红色代表最高,白色代表居中,蓝色代表最低。Heatmap图示通过Cluster和Treeviewer来进行显示。
2.8 水稻Trihelix转录因子家族的蛋白互作关系
蛋白互作分析结果显示,20个Trihelix转录因子与水稻中其他蛋白存在互作关系(图9)。从图9可以看出,Trihelix2和20,Trihelix11、12和28分别在同一个互作网络并存在直接的联系,并且Trihelix2和20位于第Ⅳ亚家族,Trihelix11、12和28位于第Ⅴ亚家族,提示其不仅在蛋白序列上具有较高同源性,同时在功能上也具有关联性。Trihelix3、5、14、23和26则只与1个其他蛋白存在互作关系,且并没有发现与Trihelix家族自身的基因存在相关性。
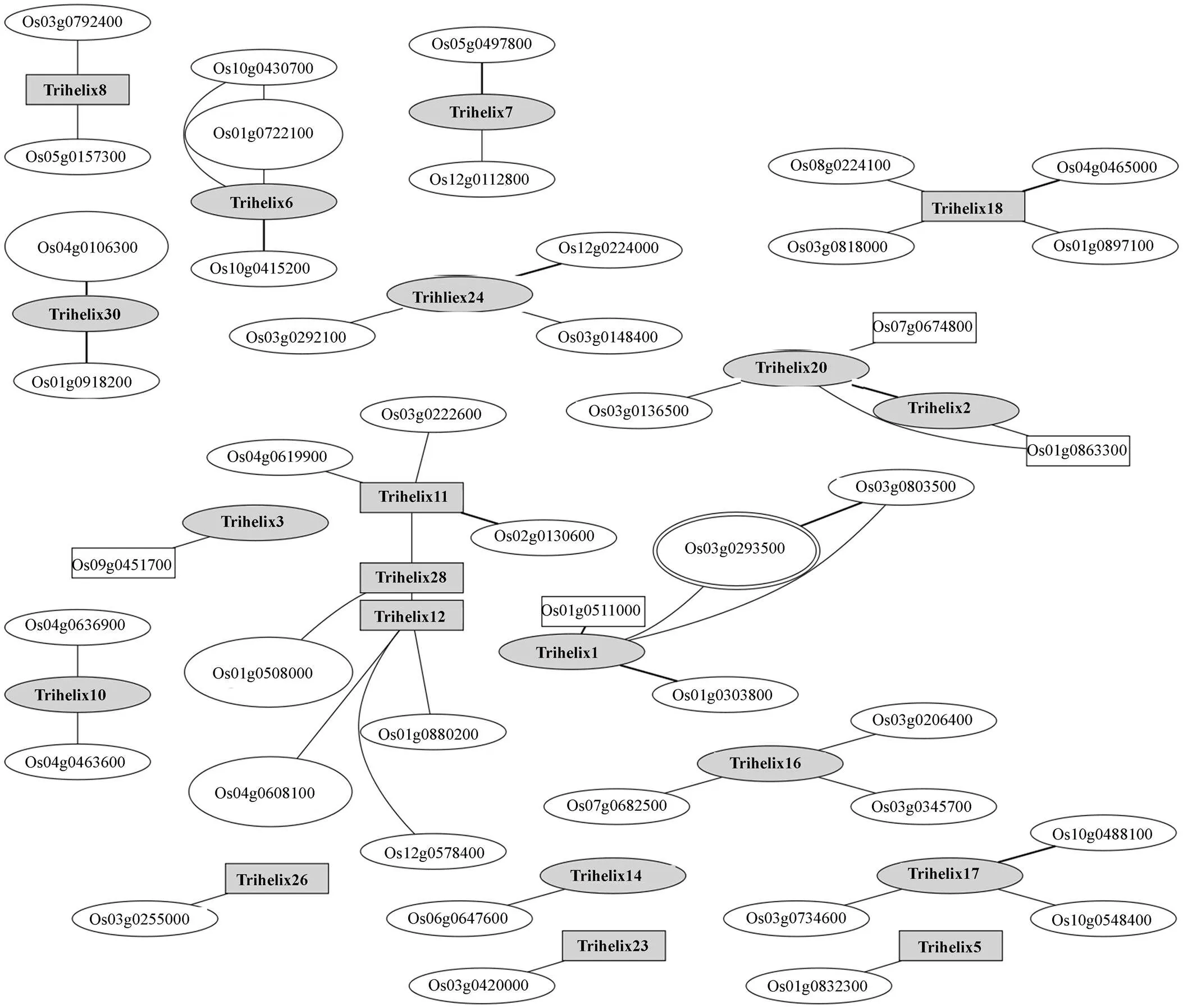
图9 Trihelix转录因子与水稻中其他蛋白的互作网络结构图
灰色框体为水稻Trihelix转录因子,空白框体为水稻中其他蛋白。
3 讨 论
相较于DOF、MYB、DREB、WRKY和NAC等转录因子家族,Trihelix是近期才被关注的转录因子家族,就基因数目而言,Trihelix只是转录因子家族中的一个小家族。Trihelix最早被发现参与光应答反应,其后被发现在植物生长发育调控以及逆境胁迫响应中起到重要作用。虽然目前已经在一些模式物种如拟南芥、大豆[31]、水稻等一些植物中对Trihelix转录因子的生物学规律开展了一些研究,并对Trihelix转录因子家族进行了初步的分类[4],但近年来随着生物信息学技术的飞速发展,利用相关的数据库和生物学软件对Trihelix转录因子家族进行更加深入和全面的分析成为可能。
本文通过对水稻Trihelix转录因子全基因组分析,将水稻Trihelix转录因子家族分为5个亚家族,结合水稻、拟南芥、二穗短炳草和高粱中Trihelix转录因子的序列分析,除Trihelix25之外,水稻Trihelix基因的亚家族划分与几个物种中的分类完全一致,表明在水稻、高粱、拟南芥和二穗短柄草发生分化之前,这些Trihelix基因已在同一物种中发生了分化。Trihelix基因的结构域的特征与Trihelix转录因子家族的分类具有一致性。在水稻Trihelix家族31个基因中,基序Motif2存在于所有基因中,Motif1则存在于除Trihelix25之外的30个基因中,结合4个物种中Trihelix基因的进化分析发现,Trihelix25独立存在于其他亚家族分区之外,这表明Trihelix25可能是进化上较为特殊的一个成员。
植物的多倍体化使基因组中保留了大量复制的染色体片段,由染色体片段复制造成基因的复制也较为常见。基因复制为新基因的产生和功能分化提供了物质基础,也为生物体的进化提供了条件。因此,研究同一物种或不同物种之间局部染色体片段的复制能够帮助人们更好地认识基因家族以及物种的进化规律。从图6可以看出,水稻4号染色体的8个基因与二穗短柄草的6个基因存在同源性,也与高粱6号染色体的7个基因存在同源性。这种线性同源性关系与水稻,二穗短柄草和高粱的基因组线性关系也保持一致[32]。其他染色体上由于基因数量较少,线性关系并不明显。
已有研究表明Trihelix转录因子在植物的营养器官和生殖器官等如花[10, 12]、气孔[33, 34]、胚胎和种子[35]的发育等不同生长发育过程中都起着重要调控作用,但在水稻中的功能研究还不多。本文利用生物芯片综合分析了Trihelix转录因子在水稻中的表达情况,并将基因表达结果聚类后分成7个不同的亚类,结果显示部分Trihelix转录因子的表达在水稻花器官相关组织中具有较高的表达并且呈现聚类现象,表现在第Ⅴ亚类的Trihelix1、2、9、20和30在子房、胚和胚乳中表达较高,第Ⅵ亚类的Trihelix5和24在花序、花药、雌蕊、外稃和内稃中表达较高,第7亚类的Trihelix11、12、19和28在外稃、内稃以及子房和胚乳的部分时期中表达较高(图7),这些分析结果将为后期研究这些基因的功能提供了一定的线索。此外,我们发现表达谱的聚类分析和蛋白聚类分析的结果虽然存在部分一致性,但并不明显,说明Trihelix转录因子的功能与其蛋白序列的相似性并不完全一致。
Trihelix转录因子除了在植物形态建成方面发挥作用之外,最近研究发现部分Trihelix基因在应对生物和非生物胁迫中扮演着重要角色[14]。本研究通过生物芯片数据分析了水稻Trihelix基因对6种植物激素的响应情况,发现不同的Trihelix基因对不同激素呈现出不同的应答模式,如Trihelix2、5、11、13、19和21对脱落酸的处理呈明显的表达上调,Trihelix1和12则相反;Trihelix1和28对细胞分裂素的处理呈明显的表达下调,Trihelix11则表现为上调。总体而言,本文所分析的6种植物激素胁迫只是逆境胁迫的一个方面,水稻Trihelix基因对更多逆境胁迫的响应机制还有待进一步研究。
参考文献
[1] Riechmann JL, Heard J, Martin G, Reuber L, Jiang C, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, Creelman R, Pilgrim M, Broun P, Zhang JZ, Ghandehari D, Sherman BK, Yu G.transcription factors: genome-wide comparative analysis among eukaryotes., 2000, 290(5499): 2105–2110.
[2] 罗军玲, 赵娜, 卢长明. 植物Trihelix转录因子家族研究进展. 遗传, 2012, 34(12): 1551–1560.
[3] Kaplan-Levy RN, Brewer PB, Quon T, Smyth DR. The Trihelix family of transcription factors-light, stress and development., 2012, 17(3): 163–171.
[4] Green PJ, Kay SA, Chua NH. Sequence-specific interactions of a pea nuclear factor with light-responsive elements upstream of the rbcS-3A gene., 1987, 6(9): 2543–2549.
[5] Perisic O, Lam E. A tobacco DNA binding protein that interacts with a light-responsive box II element., 1992, 4(7): 831–838.
[6] Le Gourrierec J, Li YF, Zhou DX. Transcriptional activation byGT-1 may be through interaction with TFIIA–TBP–TATA complex., 1999, 18(6): 663–668.
[7] Kay SA, Keith B, Shinozaki K, Chye ML, Chua NH. The rice phytochrome gene: structure, autoregulated expression, and binding of GT-1 to a conserved site in the 5' upstream region., 1989, 1(3): 351–360.
[8] Dehesh K, Bruce WB, Quail PH. A trans-acting factor that binds to a GT-motif in a phytochrome gene promoter., 1990, 250(4986): 1397–1399.
[9] Dehesh K, Hung H, Tepperman JM, Quail PH. GT-2: a transcription factor with twin autonomous DNA-binding domains of closely related but different target sequence specificity., 1992, 11(11): 4131–4144.
[10] Brewer PB, Howles PA, Dorian K, Griffith ME, Ishida T, Kaplan-Levy RN, Kilinc A, Smyth DR., a trihelix transcription factor gene, regulates perianth architecture in theflower., 2004, 131(16): 4035–4045.
[11] Lampugnani ER, Kilinc A, Smyth DR.is a boundary gene that inhibits growth between developing sepals in., 2012, 71(5): 724–735.
[12] Griffith ME, da Silva Conceicão A, Smyth DR.gene regulates initiation and orientation of second whorl organs in theflower., 1999, 126(24): 5635–5644.
[13] Lin ZW, Griffith ME, Li XR, Zhu ZF, Tan LB, Fu YC, Zhang WX, Wang XK, Xie DX, Sun CQ. Origin of seed shattering in rice (L.)., 2007, 226(1): 11–20.
[14] Fang YJ, Xie KB, Hou X, Hu HH, Xiong LZ. Systematic analysis of GT factor family of rice reveals a novel subfamily involved in stress responses., 2010, 283(2): 157–169.
[15] Barr MS, Willmann MR, Jenik PD. Is there a role for trihelix transcription factors in embryo maturation?, 2012, 7(2): 205–209.
[16] Kuromori T, Wada T, Kamiya A, Yuguchi M, Yokouchi T, Imura Y, Takabe H, Sakurai T, Akiyama K, Hirayama T, Okada K, Shinozaki K. A trial of phenome analysis using 4000-insertional mutants in gene-coding regions of., 2006, 47(4): 640–651.
[17] Kitakura S, Fujita T, Ueno Y, Terakura S, Wabiko H, Machida Y. The protein encoded by oncogene 6b frominteracts with a nuclear protein of tobacco., 2002, 14(2): 451–463.
[18] Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. Pfam: the protein families database., 2014, 42(D1): D222–D230.
[19] Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource., 2012, 40(D1): D302–D305.
[20] Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H. ExPASy: SIB bioinformatics resource portal., 2012, 40(W1): W597–W603.
[21] Jin JP, Zhang H, Kong L, Gao G, Luo JC. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors., 2014, 42(D1): D1182–D1187.
[22] Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs., 2006, 34(Suppl. 2): W369–W373.
[23] Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren JY, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching., 2009, 37(Suppl. 2): W202–W208.
[24] Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools., 1997, 25(24): 4876–4882.
[25] Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods., 2011, 28(10): 2731–2739.
[26] Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator., 2004, 14(6): 1188–1190.
[27] Lee TH, Tang HB, Wang XY, Paterson AH. PGDD: a database of gene and genome duplication in plants., 2013, 41(D1): D1152–D1158.
[28] Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics., 2009, 19(9): 1639–1645.
[29] Sato Y, Namiki N, Takehisa H, Kamatsuki K, Minami H, Ikawa H, Ohyanagi H, Sugimoto K, Itoh JI, Antonio BA, Nagamura Y. RiceFREND: a platform for retrieving coexpressed gene networks in rice., 2013, 41(D1): D1214–D1221.
[30] Zhao JH. Pedigree-drawing with R and graphviz., 2006, 22(8): 1013–1014.
[31] Guo Y, Qiu LJ. Genome-wide analysis of the Dof transcription factor gene family reveals soybean-specific duplicable and functional characteristics., 2013, 8(9): e76809.
[32] The International Brachypodium Initiative. Genome sequencing and analysis of the model grass., 2010, 463(7282): 763–768.
[33] Breuer C, Kawamura A, Ichikawa T, Tominaga-Wada R, Wada T, Kondou Y, Muto S, Matsui M, Sugimoto K. The trihelix transcription factor GTL1 regulates ploidy-dependent cell growth in thetrichome., 2009, 21(8): 2307–2322.
[34] Yoo CY, Pence HE, Jin JB, Miura K, Gosney MJ, Hasegawa PM, Mickelbart MV. TheGTL1 transcription factor regulates water use efficiency and drought tolerance by modulating stomatal density via transrepression of., 2010, 22(12): 4128–4141.
[35] Gao MJ, Lydiate DJ, Li X, Lui HL, Gjetvaj B, Hegedus DD, Rozwadowski K. Repression of seed maturation genes by a trihelix transcriptional repressor inseedlings., 2009, 21(1): 54–71.
Genome-wide analysis and functional prediction of the Trihelix transcription factor family in rice
Jianhui Ji, Yingjun Zhou, Hehe Wu, Liming Yang
The Trihelix transcription factor family plays an essential role in plant growth, development and stress response. However, the studies about identification and analysis of this gene family in rice on the genome-wide level have not been reported. In this study, 31 members of the Trihelix family, which contain highly conserved and characteristic trihelix domain through sequence clustering and functional domains analysis, were identified in rice genome database using bioinformatic tools. These members could be classified into 5 subfamilies (I~V) based on the evolutionary relationship and domain characteristics. Clustering analyses of the Trihelix family in rice,,andshowed that each species contained different members of subfamily although the classification of the Trihelix family were consistent in these four species, which indicated that the differentiation of the Trihelix gene family occur earlier than that of these species. The conserved motifs in the Trihelix family of rice analyzed using the MEME program were highly consistent with the results of clustering analyses. Intraspecific and interspecific chromosomal replication in partial Trihelix family members were found to exist in rice and between rice and other species through chromosome replication analysis. Microarray data analysis revealed diverse expression patterns of Trihelix family genes in different tissues of rice or in response to six different phytohormones. Moreover, 20 members of the Trihelix transcription factor family were found to interact with other proteins in rice using RiceFRIEND online database analysis. Therefore, our results preliminarilyidentified the evolution, chromosome distribution and replication, expression patterns, phytohormones response of the Trihelix transcription factor family andthe interaction between trihelix family proteins and other proteins in rice, which will provide a basis to further reveal the molecular evolution and biological function of the Trihelix transcription factor family.
rice; Trihelix; transcription factor family; evolution
2015-05-07;
2015-07-13
国家自然科学基金项目(编号:31400169,30900871),江苏省自然科学基金项目(编号:BK20140454,BK2011409),淮安市科技计划项目(编号:HAC2014012),江苏高校品牌专业建设工程项目(编号:PPZY2015A018)和江苏省高校“青蓝工程”项目资助
纪剑辉,讲师,研究方向:水稻分子生物学。E-mail: jijianhui@hytc.edu.cn周颖君,实验员,研究方向:植物分子生物学。E-mail: zhouzhou@hytc.edu.cn纪剑辉和周颖君并列第一作者。
杨立明,副教授,研究方向:植物抗逆分子生物学。E-mail: yanglm@hytc.edu.cn
10.16288/j.yczz.15-196
网络出版时间: 2015-11-25 17:02:28
URL: http://www.cnki.net/kcms/detail/11.1913.R.20151125.1702.002.html
(责任编委: 储成才)
猜你喜欢
南方医科大学学报(2022年3期)2022-04-13
浙江大学学报(农业与生命科学版)(2021年3期)2021-07-10
中国预防兽医学报(2020年8期)2020-11-05
三农资讯半月报(2020年15期)2020-08-25
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
上海农业学报(2017年3期)2017-04-10
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
红领巾·探索(2015年9期)2015-09-10
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
