
青霉噻唑酸表面分子印迹聚合物的制备及其吸附性能的考察
2015-12-26 01:58郑鹏磊罗智敏畅瑞苗葛燕辉
色谱 2015年9期
郑鹏磊, 罗智敏, 畅瑞苗, 葛燕辉, 杜 玮, 常 春, 傅 强
(西安交通大学药学院,陕西 西安710061)
1928 年,Fleming 发现了青霉素,随后青霉素一直广泛应用于临床,挽救了无数人的生命。青霉素具有抗菌能力强、高效、廉价等优点,然而其过敏反应的发生率居各类药物首位,被视为世界性医学难题[1,2]。近年来国内外学者研究发现致敏原并非青霉素药物本身,而是在合成、储存或使用过程中,在潮解、受热、pH 变化或酶的作用下,青霉素的β-内酰胺环易于开环产生青霉噻唑酸(penicilloic acid,PEOA)及多聚物等杂质[3]。青霉噻唑酸在体内易与蛋白质或多糖结合形成全抗原,激发T 细胞,从而诱发过敏反应,严重者甚至导致休克或死亡[4]。青霉素类抗生素被广泛应用于畜牧业,然而青霉素进入动物体内,经肝脏代谢可产生青霉噻唑酸并在肝脏蓄积,食用此类动物源食品对人们的身体健康具有潜在危害,严重影响食品的质量和安全[5]。因此建立快速有效的青霉噻唑酸分离分析方法,对提高青霉素药物的安全性、减少过敏反应的发生和检测食品中抗生素的残留有重要意义。
目前已有报道的青霉噻唑酸分析方法有薄层色谱法[6]、高效液相色谱法[7,8]、高效液相色谱-串联质谱法[9,10]、酶联免疫吸附法[11]等。此外,样品的前处理技术对于复杂样品的高灵敏度检测尤为重要,传统的前处理技术有液-液萃取、固相萃取(solid phase extraction,SPE)等,然而常用的萃取介质如C8、C18 等存在特异性识别差、回收率低、内源性杂质干扰严重等问题,导致分析结果存在重复性差、专属性不强和准确度不高的问题。近年来,为了满足高选择性的需求,新的萃取介质如分子印迹聚合物(molecularly imprinted polymers,MIPs)受到了广泛关注[12-15],分子印迹聚合物具有特异识别性、广泛实用性和构效预定性等优点,将分子印迹聚合物用于固相萃取介质可特异性识别复杂样品中的目标分子,实现对目标分子及其结构类似物的选择性富集和分离。
分子印迹聚合物的制备方法主要有本体聚合法、沉淀聚合法和悬浮聚合法等,然而这些聚合方法存在模板分子难以洗脱、印迹位点包埋过深、吸附速率慢、结合容量低等缺点[16]。近年来研究发现以表面聚合法制备的聚合物具有特异的选择性、更易接近的结合位点、快速的传质速率和较高的结合动力学等优点,表面聚合法已日益成为分子印迹领域的主要聚合技术[13,15,17]。
本研究采用表面印迹聚合法,以青霉噻唑酸为模板分子,甲基丙烯酸为功能单体,乙二醇二甲基丙烯酸酯为交联剂,甲醇/乙腈为溶剂,在改性硅胶表面制备青霉噻唑酸分子印迹聚合物,并对模板分子-功能单体摩尔比、交联度和溶剂等聚合条件进行优化。利用扫描电镜、红外光谱、元素分析和热重分析对聚合物形貌和物理特性进行表征。结合高效液相色谱法(HPLC),采用静态吸附、吸附动力学及选择性吸附实验对青霉噻唑酸分子印迹聚合物的吸附性能进行考察。所制备的分子印迹聚合物用于固相萃取(MISPE),为建立青霉素类药物和食品中致敏性杂质青霉噻唑酸的分离分析方法奠定了基础,实现青霉素药物的安全性评价和食品中青霉素残留的检测。
1 实验部分
1.1 仪器、试剂与材料
青霉噻唑酸(按照已报道的合成方法[18,19]自制);青霉素G(penicillin G,纯度>97%)、6-氨基青霉烷酸(6-aminopenicilanic acid,纯度>99%)、美洛西林(mezloeillin,纯度>98%)、氯唑西林(cloxacillin,纯度>97%)、奥拉西坦(oxiracetam,纯度>94%)均购于西安仁达生物科技有限公司;乙二酸二甲基丙烯酸酯(ethylene glycol dimethacrylate,EGDMA,美国Sigma 公司,分析纯)使用前经碱液萃取;甲基丙烯酸(methacrylic acid,MAA,天津市化学试剂研究所,分析纯)使用前经减压蒸馏处理;偶氮二乙丁腈(2,2′-azobisisobutyronitrile,AIBN,上海试剂四厂,化学纯)使用前经甲醇重结晶;3-氨丙基三乙氧基硅烷(3-aminopropyltriethoxysilane,APTES,北京百灵威化学有限公司,化学纯);磷酸氢二铵、磷酸、盐酸(北京化工厂);硅胶(粒径:100 μm,Tokyo Chemical Industry Co.,Ltd,日本);三蒸水(Molelement 1805b 元素型超纯水机,上海摩勒生物科技);其余试剂均为分析纯或色谱纯。
Shimadzu 色谱系统包括LC-20AT 泵、SPD-20A 紫外检测器、CBM-102 实时分析色谱工作站;AUY220 型电 子天平(日本 Shimadzu 公 司);Mettler Toledo pH 计(德国梅特勒-托利多集团);Thermo Nicolet Nexus 330 FT-IR Spectrometer(美国Madison 公司);TM-1000 Scanning Microscope(日本Hitachi 公司);EL3 Elementary Analyser (德国Elementar 公司);SDT Q600 Thermogravimetric Analyzer (美 国 TGA 公 司);KQ3200DB 型数控超声波清洗器(昆山市超声仪器有限公司);101-2AB 型电热恒温鼓风干燥箱(天津市泰斯特仪器有限公司)。
1.2 HPLC 条件
色谱柱:Promosil C18 柱(150 mm×4.6 mm,5 μm);流动相:乙腈-磷酸氢二铵溶液(0.05 mol/L,pH 6.18)(15 ∶85,v/v),过滤并超声后使用;流速:0.8 mL/min;检测波长:225 nm;柱温:25 ℃;进样量:10 μL。
1.3 青霉噻唑酸分子印迹聚合物的制备
采用表面印迹聚合法,以PEOA 为模板分子,MAA 为功能单体,EGDMA 为交联剂,甲醇/乙腈为溶剂,在改性硅胶表面制备青霉噻唑酸分子印迹聚合物(PEOA-MIPs)。
1.3.1 硅胶的活化
称取球形硅胶约6 g,分散于50 mL 质量分数为10% 的HCl 溶液中,在110 ℃下加热搅拌回流24 h,然后用大量水洗至中性,60 ℃下干燥24 h。
1.3.2 硅胶的改性
称取活化硅胶约5 g 于100 mL 圆底烧瓶中,依次加入甲苯50 mL、APTES 2 mL 和三乙胺1 mL,110 ℃下加热搅拌回流24 h。反应后用甲醇洗去未反应试剂,60 ℃下干燥24 h 即得改性硅胶(SiO2-APTES)。
1.3.3 PEOA-MIPs 的制备
称取0.5 mmol PEOA 于10 mL 甲醇中,超声溶解后加10 mL 乙腈,加入功能单体MAA 168 μL(PEOA 和MAA 的摩尔比为1 ∶4)和SiO2-APTES 2 g,25 ℃下搅拌12 h 进行预组装。再加入交联剂EGDMA 952 μL(交联度85%)和引发剂AIBN 0.016 4 g,超声分散20 min 后通氮气20 min,密封后于60 ℃下水浴反应24 h。将反应产物减压抽滤,并用大量甲醇洗涤,丙酮漂洗。甲醇-冰醋酸(4 ∶1,v/v)索氏洗脱24 h 以除去模板分子,然后依次用乙腈-水(20 ∶80,v/v)、甲醇洗涤,干燥得青霉噻唑酸表面分子印迹聚合物(PEOA-MIPs)。
制备青霉噻唑酸非印迹聚合物(PEOA-NIPs),除不加入模板分子PEOA 外,其他步骤同上。
1.4 聚合物形貌和物理特性的表征
采用扫描电子显微镜对活化硅胶和聚合物进行观察,以获取样品表面形貌信息。采用KBr 压片法分别对活化硅胶、改性硅胶和PEOA-MIPs 进行红外光谱分析。采用热重分析仪分别测定活化硅胶、改性硅胶及印迹聚合物的热重曲线,并根据相对重量损失计算硅胶表面的聚合物层的重量(公式(1))。对分子印迹聚合物进行元素分析,并根据元素分析结果计算聚合物层的厚度(公式(2)、(3))[20]。
其中:mMIPs为硅胶表面聚合物层的质量(mg);ms为硅胶的质量(g);loss 为由热重分析所得的质量损失;C 为聚合物层的含碳量;mC为每克硅胶表面聚合物层所含碳的质量(g);Mw为聚合物层的加权平均相对分子质量;MC为聚合物层中碳原子的加权平均相对分子质量;ρ 为加权平均密度(g/mL);S 为硅胶比表面积(m2/g);d 为聚合物层的厚度(nm)。
1.5 聚合物吸附性能的考察
1.5.1 静态吸附实验
精密称定PEOA-MIPs 和PEOA-NIPs 各20.0 mg,置于10 mL 锥形瓶中,加入10 ~800 μg/mL 的PEOA 系列溶液各10 mL,25 ℃恒温空气浴振荡1 h,吸取上清液经0.45 μm 微孔滤膜过滤后进行HPLC 分析。根据吸附前后PEOA 浓度的变化计算聚合物对PEOA 的吸附量Q(公式(4)),并用Langmuir(公式(5)[21])与Freundlich(公式(6)[22])两种模型分析静态吸附数据。

其中:C0为吸附前溶液中PEOA 的质量浓度(μg/mL);Ce为吸附后溶液中PEOA 的平衡质量浓度(μg/mL);V 为PEOA 溶液的体积(mL);W为PEOA-MIPs 或PEOA-NIPs 的质量(mg);Qe为聚合物层的吸附量(mg/g);KL为Langmuir 吸附分配系数(L/mg);Qm为聚合物层的最大吸附量(mg/g);KF和n 均为Freundlich 参数。
1.5.2 吸附动力学实验
精密称定PEOA-MIPs 和PEOA-NIPs 各20.0 mg,置于10 mL 锥形瓶中,分别加入400 μg/mL PEOA 溶液10 mL,25 ℃恒温空气浴分别振荡1、5、10、15、30、45、60、90、120 min。吸取上清液经0.45 μm 微孔滤膜过滤后进行HPLC 分析。
将吸附动力学数据用准一级动力学方程(公式(7))和准二级动力学方程(公式(8))分析[23,24]:

其中:Qe为平衡吸附量(mg/g);Qt为t 时刻的吸附量(mg/g);k1为准一级动力学方程吸附速率常数(min-1);k2为准二级动力学方程吸附速率常数(g/(mg·min))。
1.5.3 吸附选择性实验
精密称定PEOA-MIPs 和PEOA-NIPs 各20.0 mg,置于10 mL 锥形瓶中,分别加入400 μg/mL 青霉素G、6-氨基青霉烷酸、美洛西林、氯唑西林和奥拉西坦溶液各10 mL,25 ℃下恒温空气浴振摇1 h,吸取上清液经0.45 μm 微孔滤膜过滤后进行HPLC分析。
1.6 MISPE-HPLC 检测猪肝中青霉噻唑酸的前处理方法
称取150 mg PEOA-MIPs 装入2.5 mL 固相萃取空柱,柱底端和顶端均加封聚丙烯筛板,依次用6 mL 甲醇和6 mL 水冲洗。取空白猪肝脏样品2.0 g,加入4 mL 生理盐水匀浆处理,并精密吸取不同浓度青霉噻唑酸溶液200 μL 至匀浆液中,超声分散后加入6 mL 乙腈沉淀蛋白质,于8 000 r/min 下离心10 min,上清液再于12 000 r/min 下离心5 min。取上清液1 mL 上样,用1 mL 异丙醇淋洗杂质,再用3 mL 甲醇-10% 冰醋酸(9 ∶1,v/v)洗脱,收集洗脱液并用氮气吹干,用1 mL 水复溶后经0.45 μm滤膜过滤后进行HPLC 分析。
2 结果与讨论
2.1 分子印迹聚合物的制备
青霉噻唑酸分子印迹聚合物的制备流程如图1所示。硅胶颗粒经过酸活化处理,使其表面含有羟基基团。活化硅胶与APTES 发生反应,将氨基基团接枝在硅胶表面。加入的PEOA 和MAA 通过羧基和硅胶表面的氨基相互作用,从而在硅胶表面发生聚合反应,使聚合物层包覆在硅胶表面,洗脱模板后,形成青霉噻唑酸表面分子印迹聚合物。

图1 青霉噻唑酸分子印迹聚合物的制备流程Fig.1 Preparation process of PEOA-MIPs
在制备过程中功能单体MAA 可能与模板分子PEOA 形成氢键,合适的模板分子-功能单体摩尔比有利于聚合物具有更好的特异识别能力和吸附特性;交联剂使模板分子和功能单体聚合形成高度交联、刚性的聚合物,在选择交联度时,既要使聚合物具有一定的刚性以保持良好的空间构型,又要具有一定的柔韧性以便易于接近其识别位点,从而具有良好的识别性能。理想的溶剂不但有助于模板分子和其他反应物的溶解,还可能在一定程度上促进模板分子与功能单体之间的相互作用。因此为了使分子印迹聚合物具有最佳的吸附能力,分别对青霉噻唑酸分子印迹聚合物的制备条件进行了优化,结果见图2。
从图2a 中可以看出,随着MAA 的增加,PEOAMIPs 的吸附量逐渐增加;但当MAA 用量过多时,功能单体会趋于自聚合而形成较多的非特异性结合位点,导致印迹因子(IF)下降。当模板分子与功能单体的摩尔比为1 ∶4 时,所合成的聚合物有良好的识别能力与吸附能力,为最佳比例。从图2b 中可以看出,交联度为85% 时,聚合物对PEOA 的吸附量和印迹因子均相对最高,因此制备PEOA-MIPs 的最佳交联度为85%。从图2c 中可以看出,当甲醇与乙腈的体积比为3 ∶1 时,所合成的聚合物具有相对好的印迹因子,但吸附量较低;当甲醇与乙腈的体积比为1 ∶1 时,所合成的聚合物具有相对好的印迹因子和相对较高的吸附量,此时溶剂的极性有利于模板分子与功能单体之间氢键作用力的形成,因此甲醇-乙腈(1 ∶1,v/v)为最佳溶剂。综上,PEOAMIPs 的最优制备条件为PEOA/MAA 的摩尔比为1∶4,交联度为85%,溶剂为甲醇-乙腈(1 ∶1,v/v)。

图2 不同制备条件对PEOA-MIPs 吸附性能的影响Fig.2 Effect of different preparation conditions on adsorption capacity of PEOA-MIPs
2.2 聚合物形貌和物理特性的表征
2.2.1 扫描电镜分析
借助扫描电子显微镜分别对活化硅胶和聚合物的表面形貌进行了观察,结果见图3。与活化硅胶(见图3a)相比,PEOA-MIPs(见图3b)和PEOANIPs(见图3c)的表面更粗糙,表明聚合物层在硅胶表面接枝成功,而PEOA-MIPs 和PEOA-NIPs 并无明显差异。
2.2.2 热重分析

图3 (a)活化硅胶、(b)PEOA-MIPs 和(c)PEOA-NIPs的扫描电镜图Fig.3 SEM images of (a)activated silica gels,(b)PEOA-MIPs and (c)PEOA-NIPs

图4 (a)活化硅胶、(b)改性硅胶、(c)PEOA-MIPs 和(d)PEOA-NIPs 的热重分析曲线Fig.4 TGA curves of (a)activated silica gels,(b)SiO2-APTES,(c)PEOA-MIPs and(d)PEOA-NIPs
活化硅胶、改性硅胶及印迹聚合物在25 ~800℃温度区间的热重曲线见图4。随着温度的升高,活化硅胶和改性硅胶仅少量失重,在800 ℃时活化硅胶失重7.4%,改性硅胶失重10.7%,3.3% 的差额可能为所接枝APTES 的质量损失,表明氨基成功接枝在活化硅胶表面。PEOA-MIPs 和PEOANIPs 的热重曲线较为相似,在25 ~260 ℃范围内均较稳定;之后在260 ~600 ℃范围内均有较大的质量损失,随后曲线趋于平坦,在800 ℃时PEOA-MIPs和PEOA-NIPs 的总失重分别为29.0% 和38.4%,硅胶表面MIPs 层的重量约为224.0 mg/g。表明聚合物层在硅胶表面接枝成功,并且在25 ~260 ℃具有很好的热稳定性。
2.2.3 红外光谱分析
活化硅胶、改性硅胶、PEOA-MIPs 的红外光谱分析结果见图5。图5a 中3 461 cm-1处的吸收峰为活化硅胶表面O-H 的伸缩振动峰;图5b 中1 096和3 446 cm-1处为APTES 中C-N 和N-H 的伸缩振动峰,表明硅胶表面成功接枝上-NH2基团;图5c中1 109 cm-1为EGDMA 中C-O 的伸缩振动峰,1 563 cm-1处为聚合物骨架上-CH3的伸缩振动峰,1 727 cm-1处的强吸收峰可能为MAA 或EGDMA中C=O 的伸缩振动峰,相比图5b 中N-H 的伸缩振动峰变宽,说明聚合物中有氢键作用存在,结果表明聚合物成功包覆在硅胶表面。

图5 (a)活化硅胶、(b)改性硅胶和(c)印迹聚合物的红外光谱图Fig.5 FTIR spectra of (a)activated silica gels,(b)SiO2-APTESand (c)PEOA-MIPs
2.2.4 元素分析
对分子印迹聚合物进行元素分析,结果见表1。C 和N 元素在聚合物中出现,且MIPs 和NIPs 中碳的含量均较大,经计算得MIPs 和NIPs 的聚合物层厚度(d)分别约为2.17 和3.13 nm,表明功能单体和交联剂已接枝到硅胶表面,在硅胶表面附着一定厚度的聚合物。

表1 PEOA-MIPs 和PEOA-NIPs 的元素分析结果Table 1 Results of elemental analysis of PEOA-MIPs and PEOA-NIPs
2.3 聚合物吸附性能的考察
2.3.1 静态吸附实验
为考察聚合物层的吸附性能,采用静态吸附实验分别测定了PEOA-MIPs、PEOA-NIPs 和聚合物层的等温吸附曲线,结果分别见图6a、c。随着溶液中PEOA 浓度的增加,聚合物层的吸附量均增加,当溶液浓度增加至600 μg/mL,聚合物层的吸附位点趋于饱和,从而使吸附处于平衡状态,MIPs 和NIPs 聚合物层的饱和吸附量分别为122.78 和19.44 mg/g。在整个吸附过程中,PEOA-MIPs 的吸附量明显大于PEOA-NIPs,表明PEOA-MIPs 对PEOA 有特异的吸附能力。在PEOA-MIPs 制备过程中,PEOA 与MAA 之间形成氢键、静电作用力等非共价作用力,从而使在硅胶表面形成了能与模板分子PEOA 相匹配且具有特异识别位点的三维孔穴结构,因此PEOA-MIPs 除有同PEOA-NIPs 一样的非特异性吸附外,还存在对模板分子的特异性吸附位点。
将聚合物层的等温吸附数据用Langmuir 与Freundlich 两种模型进行分析,结果见表2。Langmuir 相比Freundlich 方程有更好的相关系数(R2),聚合物层对PEOA 的吸附行为符合Langmuir 模型,表明聚合物层对PEOA 的吸附是单分子层吸附。

表2 Langmuir 与Freundlich 模型的等温吸附参数Table 2 Adsorption isotherm constants for Langmuir and Freundlich equations
2.3.2 吸附动力学实验
为了研究PEOA-MIPs 达到吸附平衡所需的时间和吸附速率,PEOA-MIPs 和PEOA-NIPs 吸附动力学曲线见图6b。PEOA-MIPs 对青霉噻唑酸的吸附量在短时间内有很大的增加,在45 min 时吸附量变化趋于平缓,达到吸附平衡;且在任何时间段PEOA-MIPs 的吸附量均明显高于PEOA-NIPs。说明能与模板分子相互匹配且带有识别位点的三维空穴大都存在于聚合物的表面,因此可以快速地吸附目标化合物。
用准一级动力学方程和准二级动力学方程进行吸附动力学分析,结果见表3。准二级动力学方程的相关系数(R2)优于准一级动力学方程,聚合物对PEOA 的吸附动力学符合准二级动力学方程,表明PEOA-MIPs 吸附识别过程主要为化学吸附,PEOA-MIPs 具有快速的传质速率和结合动力学的特点。
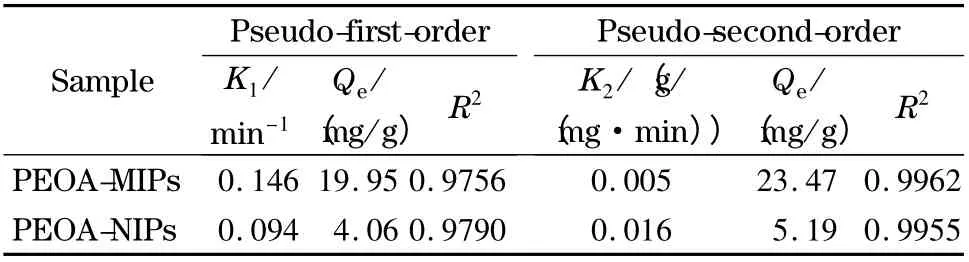
表3 准一级和准二级模型的动力学参数Table 3 Kinetic constants for pseudo-first-order rate and pseudo-second-order rate equations

图6 PEOA-MIPs 和PEOA-NIPs 的(a)等温吸附曲线、(b)吸附动力学曲线和(c)聚合物层的等温吸附曲线Fig.6 (a)Adsorption isotherms,(b)kinetics of PEOA-MIPs,PEOA-NIPs and (c)adsorption isotherms of polymer layers
2.3.3 吸附选择性实验
为考察PEOA-MIPs 对模板分子的选择性识别能力,选择6-氨基青霉烷酸、青霉素G、美洛西林、氯唑西林和奥拉西坦作为类似物进行吸附选择性实验,其化学结构和聚合物对其的吸附选择性结果见图7。PEOA-MIPs 对青霉噻唑酸的吸附能力最强,印迹因子为6.3,6-氨基青霉烷酸、青霉素G、美洛西林、氯唑西林和奥拉西坦的印迹因子分别为2.5、1.8、4.1、1.8 和1.0。在聚合物制备过程中,模板分子PEOA 在硅胶表面留下与PEOA 空间结构相匹配且具有特异识别位点的三维孔穴,因此对于非结构类似物奥拉西坦的吸附量很少,且无特异性吸附。对于6-氨基青霉烷酸、青霉素G、美洛西林和氯唑西林,结构上与青霉噻唑酸类似,存在可与PEOAMIPs 相互作用的位点,但空间结构的差异影响了聚合物的吸附能力,因此PEOA-MIPs 对其的吸附量较低,印迹因子也低于青霉噻唑酸。表明PEOAMIPs 对模板分子有高度的选择性和识别能力。
2.4 聚合方法的比较
将所得硅胶表面分子印迹聚合物与文献报道采用表面分子印迹聚合法所得的聚合物进行比较。Wei 等[25]采用溶胶-凝胶技术制备了对咖啡因具有特异识别能力的表面分子印迹聚合物,饱和吸附量为3.88 mg/g,该法制备条件温和,操作简便,但在制备过程中凝胶化的程度不易控制,溶胶-凝胶印迹层易发生收缩和干裂,导致部分结合位点被破坏,因此该法制备的聚合物存在对目标分子吸附容量低的问题,且该法制备过程耗时较长(72 h)。Li 等[26]采用表面印迹牺牲载体法制备了对盐酸小檗碱具有特异识别能力的分子印迹聚合物,所制备的颗粒形状规整,多孔性好,但当载体硅胶用氢氟酸除去后,导致聚合物机械稳定性和抗压性差,不宜用作色谱填料或固相萃取介质,且制备过程繁琐。本文利用硅胶表面的羟基基团,以APTES 修饰硅胶,从而将聚合物层嫁接在硅胶表面,操作简便,耗时较短(24 h),接枝聚合法增大了聚合物层结合位点的可接近性,使其具有更好的吸附特性,且具有机械稳定性高等优点。
2.5 样品分析
由于分子印迹聚合物对模板分子具有特异性吸附能力,因此常与固相萃取技术结合来实现对目标物质的富集、分离和纯化。本文将制备的PEOAMIPs 应用于固相萃取介质,制备了分子印迹固相萃取小柱,并与HPLC 结合,应用于猪肝中青霉噻唑酸的分离和分析。经过MISPE 净化前后样品的HPLC 色谱图见图8。经过MISPE 净化后猪肝中内源性杂质明显减少,表明MISPE 预处理可以有效地消除样品中杂质的干扰,实现对样品中PEOA 的富集和纯化。猪肝样品中PEOA 加标回收率测定结果见表4。PEOA 的回收率为87.2% ~92.3%,RSD 均小于7.9%,表明该法可用于实际样品中微量PEOA 的富集、分离和分析。

图7 PEOA 及其类似物的(a)化学结构和(b)PEOA-MIPs 对PEOA 及其类似物的吸附选择性Fig.7 (a)Chemical structures of PEOA and its analogues and (b)adsorption selectivity of PEOA-MIPs for PEOA and its analogues

图8 加标猪肝样品的色谱图Fig.8 Chromatograms of spiked pork liver samples

表4 猪肝样品中PEOA 的回收率(n=3)Table 4 Recoveries of PEOA in pork liver samples (n=3)
3 结论
以青霉素中致敏性杂质青霉噻唑酸为模板分子,采用表面分子印迹聚合法制备了PEOA-MIPs,并对制备条件进行了优化。通过SEM、FT-IR、TGA和元素分析结果显示聚合物层已成功接枝在硅胶表面,并具有良好的热稳定性。吸附性能研究发现PEOA-MIPs 对PEOA 具有良好的吸附性能和特异选择识别能力,静态吸附符合Langmuir 方程,其饱和吸附量为122.78 mg/g,并可在45 min 内达到吸附平衡,且吸附速率符合准二级动力学方程,PEOAMIPs 对目标物的吸附是以化学吸附为主的单分子层吸附。所制备的PEOA-MIPs 具有更大的吸附量、更快的吸附速率和更好的识别能力,并用于萃取介质,结合液相色谱技术实现猪肝中青霉噻唑酸的富集和分析,对于提高青霉素类药物的安全性和控制食品的质量具有重要意义。
[1] Padovan E,Bauer T,Tongio M M,et al. Eur J Immunol,1997,27:1303
[2] de Groot H,Mulder W M C. Eur J Pediatr,2010,169(11):1305
[3] Baldo B A. Antibodies,2014,3(1):56
[4] Meng X L,Jenkins R E,Berry N G,et al. J Pharmacol Exp Ther,2011,338(3):841
[5] Zhang Y,Jiang Y M,Wang S. J Agric Food Chem,2010,58:8171
[6] Moats W A. J Agric Food Chem,1983,31(6):1348
[7] Rambla-Alegre M,Martí-Centelles R,Esteve-Romero J,et al. J Chromatogr A,2011,1218(30):4972
[8] Zhang W Q,Hu Q X,Zhang X,et al. J Chromatogr A,2014,1323:87
[9] Zhang X Y,Cai X X. Chinese Journal of Chromatography(张秀尧,蔡欣欣. 色谱),2014,32(7):693
[10] Li L X,Guo C H,Ai L F,et al. J Dairy Sci,2014,97(7):4052
[11] Afolabi A S,Thottappilly G. Sci Res Essay,2008,3:524
[12] Appell M,Jackson M A,Wang L C,et al. J Sep Sci,2014,37(3):281
[13] Guijarro-Díez M,Paniagua G,Fernández P,et al. Electrophoresis,2012,33(11):1582
[14] Du W,Fu Q,Zhao G,et al. Food Chem,2013,139:24
[15] Tang K,Gu X,Luo Q,et al. Food Chem,2014,150:106
[16] Yan M. Molecularly Imprinted Materials:Science and Technology. Boca Raton:CRC Press,2004
[17] Yuan Z,Liu Y,An F. Microchim Acta,2011,172(1/2):89
[18] Isaac G S,Stanley L H,Adelbert M K. J Pharm Sci,1984,73(1):125
[19] Luo Z M,Zhou H Y,Wang W W,et al. Chinese Journal of Pharmaceutical Analysis (罗智敏,周会艳,王薇薇,等. 药物分析),2013,33(4):628
[20] Halhalli M R,Schillinger E,Aureliano C S A,et al. Chem Mater,2012,24(15):2909
[21] Dada A O,Olalekan A P,Olatunya A M,et al. J Appl Chem,2012,3(1):38
[22] Mazzotti M. J Chromatogr A,2006,11(26):311
[23] Lee C R,Kim H S,Jang I H,et al. ACS Appl Mater Inter,2011,3(6):1953
[24] Ho Y S,McKay G. Process Biochem,1999,34:451
[25] Wei H S,Tsai Y L,Wu J Y,et al. J Chromatogr B,2006,836:57
[26] Li H,Li Y,Li Z,et al. Appl Surf Sci,2012,258(10):4314
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
云南画报(2021年10期)2021-11-24
小学生优秀作文(高年级)(2018年4期)2018-09-11
郑州大学学报(工学版)(2015年1期)2015-03-24
中国当代医药(2015年26期)2015-03-01
现代检验医学杂志(2015年4期)2015-02-06
现代检验医学杂志(2015年2期)2015-02-06
中国摄影(2014年12期)2015-01-27
食品工业科技(2014年9期)2014-03-11
中国兽药杂志(2012年4期)2012-11-06
