
稳定同位素编码衍生-分散液液微萃取-超高效液相色谱-三重四极杆质谱检测大鼠脑微透析液中左旋多巴和多巴胺
2015-12-26 01:58亓伟梅赵先恩孙志伟尤进茂索有瑞
色谱 2015年9期
亓伟梅, 赵先恩 , 亓 永, 孙志伟, 陈 光, 尤进茂,* , 索有瑞
(1. 莱芜职业技术学院化工教研室,山东 莱芜271100;2. 山东省生命有机分析重点实验室,山东省高校绿色天然产物与医药中间体重点实验室,曲阜师范大学化学与化工学院,山东 曲阜273165;3. 中国科学院西北高原生物研究所,青海 西宁810001)
左旋多巴(L-dihydroxyphenylalanine,LDOPA)是多巴胺(dopamine,DA)体内合成的前体物质,主要通过血脑屏障转运入脑,经体内代谢转化为药效成分DA,从而发挥其“替代疗法”作用,是目前临床方面改善帕金森病(PD)症状最有效的药物之一。但是,长期使用L-DOPA 疗效会降低且会给病人带来严重的副作用,包括症状波动、运动障碍、开-关反应及精神症状等,给PD 中晚期的治疗带来严重的困难。已有研究报道L-DOPA 副作用的产生与其脑内浓度、所转化的DA 浓度波动具有密切相关性[1-3]。因此,同时监测脑内L-DOPA 和DA浓度的动态变化,能够为提高L-DOPA 对PD 的临床疗效以及药物筛选提供重要依据。
已报道的L-DOPA 和DA 的检测方法有荧光光度法、化学发光法、电化学法、HPLC、CE、LC-MS等[4-6]。因为脑微透析液样本量少、L-DOPA 和DA含量低且基质复杂,直接检测时荧光或质谱响应信号弱。衍生化LC-MS/MS 法因其选择性、灵敏度、抗基质干扰、多成分同时检测等优势成为一类重要方法[4,7-10]。最近,稳定同位素衍生化LC-MS/MS法相继被报道,结果表明其具有较好的灵敏度和抗基质干扰能力[11-15]。同时,超声辅助-分散液液微萃取(ultrasonic-assisted dispersive liquid-liquid microextraction,UA-DLLME)联合衍生化的样品前处理技术已在分析化学的交叉学科研究中展现出明显的优势[16-19]。
本文采用d0/d3-10-甲基-吖啶酮-2-磺酰氯(d0/d3-MASC)作为稳定同位素衍生化试剂[13],联合UA-DLLME 技术,建立并验证了帕金森病大鼠脑微透析液中L-DOPA 和DA 的超高效液相色谱-三重四极杆质谱(UHPLC-MS/MS)检测方法,为左旋多巴的临床疗效及减毒增效药物筛选提供了一种良好的技术手段,具有活体、连续、灵敏、快速、准确等特点。
1 实验部分
1.1 仪器与试剂
美国Waters Xevo TQ 三重四极杆质谱仪(Waters公司);瑞典CMA 微透析取样系统,包括CMA 402 双通道微透析泵、MAB85 双通道冷却收集器、MAB6 脑探针及导引管,以及动物五通道清醒活动装置(美国Instech 公司);动物手术设备:CMA450 动物体温控制器、美国ASI SAS-4100 单臂脑立体定位仪、颅钻(深圳沃瑞德科技有限公司);BS l10S 分析天平(北京赛多利斯有限公司);Milli-Q 超纯水仪;Eppendorf 5810R 高速冷冻离心机(德国Eppendorf公司);超声波清洗器(昆山超声仪器公司);XW-80A 漩涡混匀器(上海精科实业有限公司);雷兹pH 计。
L-DOPA 和DA(Sigma 公司);乙腈、甲醇、甲酸均为色谱纯(Sigma 公司);碳酸钠、碳酸氢钠等为分析纯;同位素衍生化试剂d0/d3-MASC(纯度99%)自主合成[13];水为Millipore 纯水系统制备。
1.2 溶液配制
按实验要求称取适量d0/d3-MASC,用乙腈配成1.0×10-3mol/L 的溶液。分别称取适量L-DOPA和DA 标准品,用50% (v/v)乙腈水溶液配成0.01 mol/L 储备液,稀释 得系 列 工 作 液。Na2CO3-NaHCO3缓冲 液(0.1 mol/L)经 纯 水 配 制 后 用NaOH 溶液精密调节pH 至10.8。
1.3 活体大鼠脑微透析取样及动物分组
雄性SD 大鼠(200 ~220 g),购自山东鲁抗医药有限公司实验动物中心,动物手术、PD 动物模型和微透析探针手术技术按照王丹巧研究组[3]报道的方法开展实验,脑内注射6-羟基多巴胺(6-OHDA)造模,分为正常组、PD 模型组、PD +L-DOPA组、PD+L-DOPA+首乌方高剂量组(PD+L-DOPA+high SWF,生药量18 g/(kg·d))、PD+L-DOPA+首乌方低剂量组(PD+L-DOPA+low SWF,生药量8 g/(kg·d)),每组6 只。微透析灌 流 速 度2.0 μL/min,平衡60 min 后开始收集透析液,每15 min自动收集1 管用于后续检测。
1.4 同位素编码衍生
在1.5 mL 离心管中加入100 μL d0-MASC 溶液、适量混合标准溶液(或大鼠脑微透析液样本,20~50 μL)、100 μL Na2CO3-NaHCO3缓冲液,混匀后37 ℃下恒温衍生反应3.0 min 即可完全标记,加入10 μL 25% (v/v)甲酸水溶液调pH 至弱酸性。在第二支离心管中,采取同样的方式,用d3-MASC 标记另一份混合标准溶液或微透析液样品,标记流程示意图见图1。

图1 L-DOPA 和DA 的稳定同位素编码衍生和分散液液微萃取示意图Fig.1 Procedure scheme of stable isotope-coded derivatization and dispersive liquid-liquid microextraction of L-DOPA and DA
1.5 超声辅助-分散液液微萃取
将上述两支离心管中的标记产物合并至一支10 mL 尖底离心管中,加水至8 mL,以400 μL 甲醇为分散剂,160 μL 氯仿为萃取剂,室温下超声振荡萃取3.0 min,10 000 r/min高速离心3.0 min,吸取下层有机相,氮吹至干,50% (v/v)甲醇水复溶后用于检测。
1.6 色谱-质谱条件
色谱柱为UPLC BEH Shield RP 18 柱(50 mm×2.1 mm,1.7 μm,Waters 公司)。流动相A 为甲醇,流动相B 为0.1% (v/v)甲酸水溶液。梯度洗脱条件:0 ~2.0 min,65% A ~100% A。流速0.5 mL/min,柱温30 ℃,进样量5 μL。采用三重四极杆质谱的多反应监测(MRM)模式,电喷雾电离(ESI)源正离子模式,离子源温度150 ℃,毛细管电压3.0 kV,脱溶剂气(氮气)流速800 L/h、温度350℃,锥孔气(氮气)流速50 L/h,碰撞气(氦气)流速0.16 mL/min,锥孔电压、碰撞能、定量和定性离子对参数见表1。
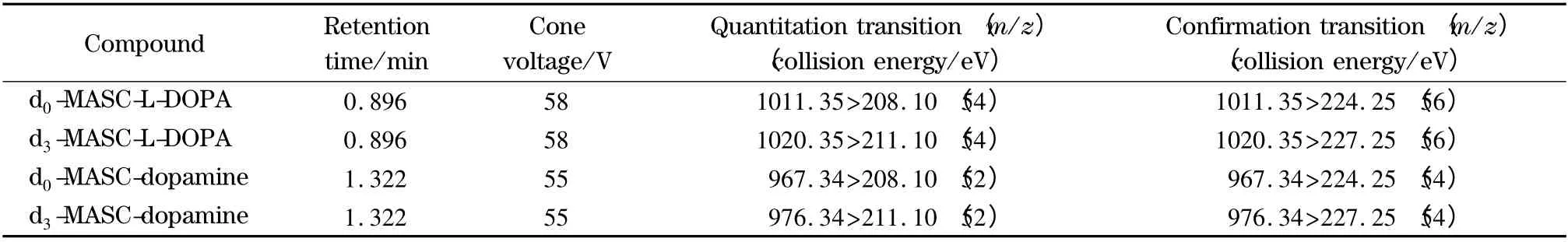
表1 L-DOPA 和DA 的d0/d3-MASC 衍生物的质谱MRM 采集参数Table 1 Conditions of d0/d3-MASC derivatives of L-DOPA and DA in mass spectrometric MRM analysis
1.7 定量方式
参考许国旺研究组[20]和本研究组[15]的同位素编码标记定量法:两份等浓度混合标准溶液分别用d0-MASC 和d3-MASC 标记,将两份衍生溶液按体积比1/10、1/5、1/2、1/1、2/1、5/1、10/1 混合之后进行UHPLC-MS/MS 分析,以轻/重衍生化产物的质谱峰面积比值对体积比值进行线性回归曲线制作,获得轻/重衍生物的相对响应因子。微透析液和适宜浓度的标准溶液经轻/重编码标记后,按一定比例混合之后进行UHPLC-MS/MS 分析,利用相对响应因子计算微透析液中两种分析物的含量。
2 结果与讨论
2.1 色谱-质谱条件的优化
L-DOPA 和DA 分子本身极性强,采用直接LCMS 检测时色谱保留弱、分离度较差,采用本研究实验部分梯度洗脱条件时,二者的保留时间均小于0.5 min。采用MASC 衍生化技术,能够显著改善二者在反相色谱柱上的保留行为。经优化不同色谱柱(Agilent 公司的Eclipse、Zorbax 系列的50、100 mm 长的超高效液相色谱柱,以及Waters BEH 系列的50 mm 超高效液相色谱柱)和流动相条件(甲醇、乙腈、水相是否有甲酸调节剂),发现酸性的甲醇/水体系配合Waters UPLC BEH Shield RP 18 超高效液相色谱柱有较好的分离效果。如要在1.0 min 左右完成分离检测,两种分析物的峰形、分离度欠佳,影响定量准确度。进一步优化梯度洗脱程序,当采用1.6 节中梯度洗脱程序时,L-DOPA 和DA衍生物在2.0 min 内实现了完全分离,峰形良好,保留时间见表1。经实验对比发现,正离子模式的灵敏度显著高于负离子模式。在正离子模式下,发现毛细管电压在1.2 ~3.0 kV 内逐渐增加时检测信号逐渐增强,实验选用3.0 kV。质谱仪脱溶剂气流速和温度影响不明显,采用脱溶剂气流速800 L/h、温度350 ℃能获得较好的实验结果。采用实验部分的质谱条件,优化了两种衍生产物的MRM 参数(见表1)。L-DOPA 和DA 衍生物都能特异性地产生208.10 Da (重标211.10 Da)的产物离子,在酸性乙腈/水流动相体系中,该产物离子结构通过酮式-烯醇式互变,极易产生带一个正电荷的季铵型结构,因此产生L-DOPA 和DA 吖啶酮衍生物的质谱增敏结果。代表性的d0-MASC-L-DOPA 衍生物的MS/MS 图、裂解模式及质谱增敏产物离子见图2。
2.2 衍生条件的优化
采用MASC 试剂标记L-DOPA 和DA 分子中的氨基和酚羟基,该衍生化反应的主要影响因素包括缓冲溶液pH、衍生化温度、时间、衍生化试剂倍数。参照以往MASC 衍生化环境雌激素分子中酚羟基基团的方法与条件[13]进行单因素法优化。首先考察缓冲液pH 在8.0 ~11.0 范围内对衍生反应的影响,pH 8.0 ~9.0 时目标衍生化产物的仪器信号极低;pH>9.0 时信号随pH 增大而逐渐提高;pH 10.8时达到最大值,之后仪器信号下降,这可能是因为强碱性导致衍生试剂和衍生物的分解更甚。因此实验选用pH 10.8 的碳酸钠-碳酸氢钠缓冲液。同样考察MASC 用量、衍生时间、温度变化规律。随MASC用量增加,衍生物的仪器信号逐渐增大;当MASC用量为两种分析物总物质的量的8 倍时仪器信号最高。在衍生时间和温度方面,37 ℃下衍生3 min 产物的信号最大。总之,实验最佳衍生化条件为:Na2CO3-NaHCO3缓冲液(pH 10.8),37 ℃下衍生反应3 min,MASC 物质的量为两种分析物总量的8 倍。

图2 d0-MASC-L-DOPA 的MS/MS 图、特异性产物离子裂解模式及MS 增敏机理Fig.2 MS/MS spectrum,specific fragmentation scheme and MS sensitivity enhancement mechanism of d0-MASC-L-DOPA
2.3 UA-DLLME 条件的优化
UA-DLLME 实验的主要影响因素包括萃取剂、分散剂的种类、用量以及超声时间。经实验优化发现,萃取剂、分散剂的种类和用量是影响萃取效率的最重要因素。选取5 种萃取剂(氯苯、二氯乙烷、四氯化碳、三氯甲烷、二氯甲烷)和4 种分散剂(甲醇、乙醇、乙腈、丙酮)进行分别交叉组合考察。实验结果表明,三氯甲烷作为萃取剂、甲醇作为分散剂时,不但易于离心分层,而且沉积相中萃取物的仪器响应信号最高,且分析方法回收率较好。因此,实验选择三氯甲烷作为萃取剂,甲醇作分散剂。考察萃取剂和分散剂的用量,当三氯甲烷160 μL、甲醇400 μL 时,沉积相中萃取物的仪器信号响应值最高。采用以上最佳条件进行超声萃取时间的优化,发现超声振荡3.0 min 可获得最高的仪器响应值。采用以上优化条件进行一次UA-DLLME 后,吸净下层沉积相,再对上层溶液重复进行一次UA-DLLME 和检测,未检出L-DOPA 和DA 衍生物。实验结果表明,建立的UA-DLLME 条件对L-DOPA 和DA 衍生物的萃取效率满足分析方法要求。
2.4 分析方法的评价
2.4.1 线性关系、检出限及定量限
采用优化的衍生化、UA-DLLME 和色谱-质谱分析条件及定量方式,在0.20 ~1 500.0 nmol/L 浓度范围内进行线性回归曲线制作,L-DOPA和DA 衍生物的线性回归系数大于0.994,线性关系良好。L-DOPA、DA 的检出限(S/N =3)分别为0.005、0.009 nmol/L,定量限(S/N =10)分别为0.020、0.036 nmol/L。
2.4.2 稳定性、精密度和准确度
在上述实验条件下,在PD 症大鼠脑微透析液中加标浓度分别为0.10、10.0 nmol/L 时制备MASC 的衍生物,用于稳定性、精密度及准确度的考察。上述两个浓度水平的混合标准MASC 衍生化产物溶液在室温下放置7 天,分别于第1、3、5、7 天测定峰面积,计算峰面积的RSD,在4.1% ~8.9%之间,表明L-DOPA 和DA 的MASC 衍生产物稳定性良好,能够满足实验要求。同一日内连续进样5次、连续3 日每日进样5 次分别用于日内和日间精密度、准确度的考察,峰面积的RSD 为3.0% ~9.1%,精密度良好;添加物的检出浓度和理论浓度比值在89.5% ~104.9% 之间,准确度良好。
2.4.3 基质效应和回收率
考察PD 症大鼠脑微透析液在0.10、10.0 nmol/L 加标水平下的回收率,结果为94.8% ~103.8% ,表明方法回收率良好。按照(微透析液中添加的L-DOPA 或DA 质谱检测峰面积/纯水中等量添加物峰面积)×100% 计算基质效应,结果为97.5%~103.8% ,表明同位素编码衍生化与DLLME 联合使用的方法具有良好的抗基质干扰能力。
2.5 方法比较
用本方法与神经递质分析方法领域的代表性研究[7,10,20-22]进行比较。第一,与2 种未经衍生的代表性的直接检测方法HPLC-电化学法[20]和亲水作用色谱-质谱(HILIC-MS/MS)[21]相比,本方法中两种分析物的LOD 是其1/55 ~1/10 944 ,分析时间短10 或18 min。第二,与3 种典型的衍生化LCMS/MS 检测方法相比,本方法的LOD 是文献[7,10]的1/3 ~1/80,LOQ 是文献[22]的1/1 265 ,而且本方法的衍生化反应温度适合生物样本,衍生化时间缩短为原来的1/8 ~1/20,分离时间与文献相当[10,22]或更短[7]。第三,在基质效应方面,用本方法与不采用同位素编码衍生技术(具体方法如下:只使用d0-MASC 衍生,UA-DLLME 和质谱分析基本条件与本方法相同,定量方式采用文献[17]中常规的外标法)进行对比,结果表明,本方法基质效应的平均值为98.9%、标准偏差为3.4%,显著优于只用d0-MASC 衍生的实验结果(基质效应的平均值为94.6%、标准偏差为11.8%),这说明同位素编码衍生化联合UA-DLLME 技术能够显著降低基质干扰。
2.6 首乌方对PD 大鼠脑微透析液中L-DOPA 和DA 浓度的影响
王丹巧研究组[3]采用PD 大鼠模型考察了中药方剂首乌方协同的L-DOPA 药代动力学,发现在以较小剂量L-DOPA 发挥较大药效作用方面,中药方剂首乌方具有良好的协同作用,但该报道未同时检测DA 浓度受到的影响。本文利用该动物模型,进一步考察高、低剂量的首乌方与L-DOPA 同时使用时对大鼠脑内纹状体L-DOPA 和DA 浓度的影响。按照实验部分的步骤,将采集得到的各组大鼠脑微透析液样本按照本法进行检测,结果表明,PD 模型组大鼠纹状体DA 含量显著低于正常组(p<0.01),从生化分析层面证明本研究PD 动物模型复制成功。表2 为3 组PD 症大鼠给予西药、中西药结合(首乌方高、低剂量组)干预后的脑纹状体L-DOPA和DA 的浓度结果。正常组、PD 模型组、PD +LDOPA+首乌方高剂量组的大鼠脑微透析液样品UHPLC-MS/MS (MRM)分析结果见图3,峰形、分离度良好,未发现干扰物质。实验结果表明,与PD+L-DOPA组相比,PD+L-DOPA+首乌方高、低剂量组纹状体细胞外液中L-DOPA 和DA 二者的浓度达到的最大值更低,且时间滞后,而且可以稳定纹状体L-DOPA 药物及活性成分DA 的浓度,使脑内二者浓度较长时间维持在有效水平,这些实验结果与首乌方高、低剂量具有相关性。这提示首乌方中的中药成分可延缓L-DOPA 的代谢和消除,减小它们的浓度波动范围,以提高L-DOPA 的疗效,减少副作用,该实验结果与文献[3]中探讨的首乌方协同LDOPA 治疗PD 的机制相同。

表2 L-DOPA 和DA 在大鼠纹状体微透析液中浓度的动态变化(n=6)Table 2 Dynamic concentration changes of L-DOPA and DA in rat striatum microdialysate (n=6)

图3 大鼠脑微透析液中L-DOPA 和DA d0/d3-MASC 衍生物的UHPLC-MS/MS (MRM)色谱图Fig.3 UHPLC-MS/MS (MRM)chromatograms of L-DOPA and DA derivatives of d0/d3-MASC in rat brain microdialysate
3 结论
建立并验证了同时测定L-DOPA 和DA 的稳定同位素编码衍生-分散液液微萃取UHPLC-MS/MS分析方法,该方法具有灵敏、准确、快速、专属性强、抗基质干扰等特点,能有效监控不同组PD 大鼠脑微透析液中L-DOPA 药物和生物活性成分DA 的含量,为PD 相关的医学检验和药物筛选提供了一种技术手段。
[1] Zhou H H. Pharmacology. Beijing:Science Press (周宏灏.药理学. 北京:科学出版社),2007:115
[2] Wang D Q,Wang W,Jing F C,et al. Chinese Pharmacological Bulletin (王丹巧,王巍,景富春,等. 中国药理学通报),2007,23(11):1527
[3] Sun X F,Wang D Q,Wu Z E,et al. Chinese Journal of Experimental Traditional Medical Formulae (孙晓芳,王丹巧,吴兆恩,等. 中国实验方剂学杂志),2011,17(11):111
[4] Perry M,Li Q,Kennedy R T. Anal Chim Acta,2009,653:1
[5] Zhao Y Y,Liu L Y,Han Y Y,et al. Chinese Journal of Chromatography (赵燕燕,刘丽艳,韩媛媛,等. 色谱),2011,29(2):146
[6] Song Y Q,Li F Y,Li W,et al. Chinese Journal of Health Laboratory Technology (宋彦强,李凤云,李玮,等. 中国卫生检验杂志),2013,23(5):1339
[7] Song P,Mabrouk O S,Hershey N D,et al. Anal Chem,2012,84:412
[8] Zhao X E,Suo Y R. Talanta,2008,76:690
[9] Zhao X E,Suo Y R. Chinese Journal of Analytical Chemistry(赵先恩,索有瑞. 分析化学),2008,36(1):12
[10] Ji C J,Li W L,Ren X D,et al. Anal Chem,2008,80:9195
[11] Liu P,Huang Y Q,Cai W J,et al. Anal Chem,2014,86:9765
[12] Leng J P,Wang H Y,Zhang L,et al. Anal Chim Acta,2013,758:114
[13] Zhang S J,You J M,Ning S J,et al. J Chromatogr A,2013,1280:84
[14] Dai W D,Huang Q,Yin P Y,et al. Anal Chem,2012,84:10245
[15] Sun Z W,Wang X X,Cai Y P,et al. Talanta,2014,120:84
[16] Yuan K,Kang H N,Yue Z F,et al. Anal Chim Acta,2015,866:41
[17] Zhao X E,Lü T,Wei N,et al. Chinese Journal of Analytical Chemistry (赵先恩,吕涛,魏娜,等. 分析化学),2014,42(11):1629
[18] Wang X Y,Qi W M,Zhao X E,et al. Chinese Journal of Chromatography (王晓燕,亓伟梅,赵先恩,等. 色谱),2014,32(6):623
[19] Wu C Q,Lei J M,Li Y L,et al. Chinese Journal of Chromatography (吴翠琴,雷金妹,李韵灵,等. 色谱),2014,32(12):1362
[20] Wu Z E,Niu X H,Zhang M Y,et al. Chinese Journal of Pharmaceutical Analysis (吴兆恩,牛晓红,张美玉,等. 药物分析杂志),2012,32(2):217
[21] Tufi S,Lamoree M,Boer J D,et al. J Chromatogr A,2015,1395:79
[22] Junnotula V,Licea-Perez H. J Chromatogr B,2013,926:47
猜你喜欢
中国现代药物应用(2022年16期)2022-09-29
中国实用神经疾病杂志(2021年16期)2021-01-03
世界最新医学信息文摘(2020年23期)2020-12-25
祝您健康(2018年8期)2018-08-18
实用心脑肺血管病杂志(2018年4期)2018-06-12
中成药(2018年4期)2018-04-26
中成药(2017年4期)2017-05-17
中国医学创新(2016年29期)2016-02-17
中国继续医学教育(2015年3期)2016-01-06
肾脏病与透析肾移植杂志(2014年6期)2014-04-05
