
非血红素铁模型配合物[FeⅣ(O)(N4Py)]2+的磺化氧化反应机理
2016-06-22 06:24刘媛,王一,刘文华,唐喆,张爱杰
大连工业大学学报 2016年3期
刘 媛, 王 一, 刘 文 华, 唐 喆, 张 爱 杰
( 1.大连工业大学 生物工程学院, 辽宁 大连 116034;2.大连工业大学 机械工程与自动化学院, 辽宁 大连 116034;3.山西师范大学 化学与材料科学学院, 山西 临汾 041001 )
非血红素铁模型配合物[FeⅣ(O)(N4Py)]2+的磺化氧化反应机理
刘 媛1,王 一1,刘 文 华2,唐 喆1,张 爱 杰3
( 1.大连工业大学 生物工程学院, 辽宁 大连116034;2.大连工业大学 机械工程与自动化学院, 辽宁 大连116034;3.山西师范大学 化学与材料科学学院, 山西 临汾041001 )
摘要:利用密度泛函理论研究了4价非血红素铁模型配合物[FeⅣ(O)(N4Py)]2+的磺化氧化反应机理。选取苯硫基甲烷及其对位衍生物为反应底物,苯硫基甲烷对位取代基的吸电性越强,硫原子的Mulliken电荷数就越大。磺化氧化反应的反应机理是在反应过程中发生了直接的两电子转移。氧转移反应为两态反应,反应活性主要由五重态的反应能垒决定。通过计算可知,对位取代基的吸电性越强,其反应能垒就越高,反应活性顺序由高到低依次为对甲基苯硫基甲烷、苯硫基甲烷,对氯苯硫基甲烷、对氰基苯硫基甲烷、对硝基苯硫基甲烷。
关键词:非血红素;磺化氧化反应;密度泛函理论;氧转移反应
0引言
含有单核非血红素铁活性中心的氧激活酶可以参与许多重要的反应,在环境、生理和药物开发等领域具有重要的地位和作用[1]。单核非血红素铁氧活化酶催化反应中间体通常是高价铁氧配合物,可以参与许多重要的催化循环过程[2]。2003年,Que等首次合成可以独立并稳定存在的6齿配位4价铁氧配合物[3],随后,合成了以N4和N4S为配位基的4价铁氧配合物[4-6],其性质可以由莫斯堡谱[7-9]、拉曼光谱[10]、X衍射[1-12]和理论计算[13-15]等手段获得。在生物合成领域,4价铁氧配合物的反应活性和稳定性与配位基的结构有着很大关系。非血红素铁模型配合物[FeⅣ(O)(N4Py)]2+的赤道方向有4个吡啶配位基,这4个配位基的方向都与FeO轴平行。[FeⅣ(O)(N4Py)]2+具有热力学稳定性,在25 ℃时,半衰期为60h,X衍射晶体结构数据显示Fe—O键长为0.163 9nm[4]。大量的实验数据[4,8]表明,[FeⅣ(O)(N4Py)]2+有着强氧化能力,不仅可以发生氢夺取反应,还可以发生氧转移反应。它具有活化强C—H键的能力,甚至可以活化环己烷的C—H键(DCH=414kJ/mol)。KumarD[16]利用密度泛函理论研究了[FeⅣ(O)(N4Py)]2+与环己烷的羟基化反应。研究结果发现,羟基化反应是两态反应,其速控步骤是氢夺取反应,其羟基化反应的反应活性甚至强于细胞色素P450。
尽管细胞色素P450的磺化氧化反应已有广泛研究[17-19],但非血红素铁模型配合物的磺化氧化反应的系统研究还比较少。[FeⅣ(O)(N4Py)]2+可以发生磺化氧化反应,其氧化苯硫基甲烷的速率常数k2为0.065mol/s[14]。高价金属氧化物发生磺化氧化反应通常有两种反应机理[17,20]:直接的氧转移反应(DOT);先发生电子的转移,然后是氧转移反应(ETOT)。为了能更好地探究其反应机理,本文利用密度泛函理论研究了[FeⅣ(O)(N4Py)]2+氧化苯硫基甲烷及其对位衍生物的反应机理。苯硫基甲烷在常温常压下性质稳定,常被用作有机合成的中间体。其结构中含有苯环,可发生亲核取代和亲电取代反应,通过选取具有不同性质的对位取代基,可以更系统地研究4价非血红素铁模型配合物的磺化氧化反应的反应机制。
1计算方法
本文选取的模型是六齿配位的配合物[FeⅣ(O)(N4Py)]2+。全部计算使用Gaussian09软件包。泛函为非限制性的B3LYP[21-23]。所有的优化与反应通道的计算都在溶剂里进行,其模型为自洽反应场的连续极化模型[24],溶剂选取的是乙腈。几何构型的优化都没有限制其对称性。对于铁原子,采用的是LACVP基组,C、H、N、O、S、Cl采用的是6-31G**基组[25-26](简称为B1)。单点能计算,所有的原子都采用def2-TZVP[27]基组(简称为B2//B1)。对于苯硫基甲烷及其对位衍生物,利用UB3LYP/TZVP[28-29]对其进行优化。
2结果与讨论
2.1[FeⅣ(O)(N4Py)]2+和苯硫基甲烷及其衍生物的电子结构和几何构型
图1为[FeⅣ(O)(N4Py)]2+的几何结构,以及在UB3LYP/B2//B1和UB3LYP/B2//B1+ZPE计算水平下,单重态、三重态和五重态的相对能量。通过计算结果可知,三重态为基态。三重态时,配合物的Fe—O键长为0.162 7nm,Fe—Nax的键长为0.207 5nm,五重态时分别为0.162 2和0.209 6nm。[FeⅣ(O)(N4Py)]2+的三重态和五重态变化最大的就是Fe与赤道方向N的平均键长,三重态时Fe—Neq为0.198 8nm,五重态时为0.211 8nm。

图1[FeⅣ(O)(N4Py)]2+单重态(三重态)[五重态]的几何结构和相对能量
Fig.1The optimized structures and the relative energies of [FeⅣ(O)(N4Py)]2+for the singlet (triplet) [quintet] spin states
图2为UB3LYP/TZVP计算水平下苯硫基甲烷及其对位衍生物的几何和电子结构。5个反应底物分别是p-Me-thioanisole、thioanisole、p-Cl-thioanisole、 p-CN-thioanisole和p-NO2-thioanisole。氢原子、氯原子、氰基、硝基都是吸电子取代基,电负性越大吸引电子的能力越强,其电负性大小的顺序为—NO2、—CN、Cl、H,而甲基为斥电子取代基。由图2可知,对位取代基的电负性越大,α-HOMO的轨道能量就越低,硫原子的Mulliken电荷也就越大(值越正),C—S的键长也就越短。
2.2磺化氧化反应的反应机理
图3给出了在B2//B1计算水平下,[FeⅣ(O)(N4Py)]2+与苯硫基甲烷及其对位衍生物的反应能垒。反应由三重态开始,经过五重态过渡态,最后形成产物。相对于3RC,五重态的反应能垒分别为44.4、47.3、48.5、52.3和53.1kJ/mol。计算结果表明衍生物对位取代基的吸电性越强,反应能垒就越高。

图2UB3LYP/TZVP计算水平下苯硫基甲烷及其对位衍生物的几何和电子结构(QS为硫原子的Mulliken电荷)
Fig.2Key geometric and electronic features of UB3LYP/TZVP-optimized five substrates (QS:Mulliken charges of S)
如图3所示,五重态过渡态时,Fe—O的键长分别为0.164 0、0.164 2、0.164 6、0.165 7和0.166 7nm,S—O键的键长分别为0.292 2、0.287 5、0.280 6、0.267 1和0.257 8nm。由此说明对位取代基的吸电性越强,Fe—O键的键长越长,而S—O的键长越短,说明过渡态形成的越晚,这与反应能垒的趋势相一致。


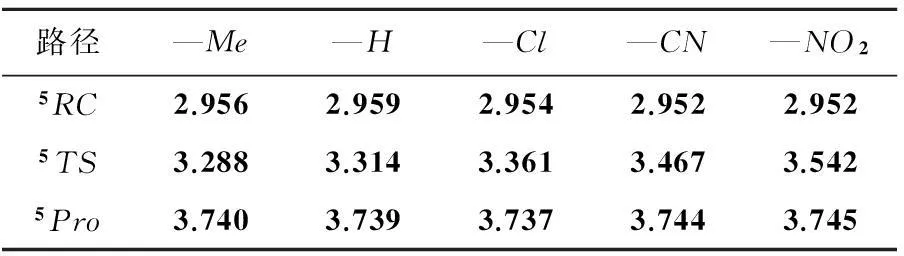
表1 Fe原子在反应过程中的电子结构参数
3结论
4价非血红素铁模型配合物[FeⅣ(O)(N4Py)]2+的基态为三重态。苯硫基甲烷对位取代基基团的吸电性越强,硫原子的Mulliken电荷就越正。计算结果显示,相对于3RC,五重态的反应能垒分别为44.4、47.3、48.5、52.3和53.1kJ/mol,也就是说苯硫基甲烷对位取代基的供电性越强,其反应就越难发生。同时研究了过渡态的几何结构,Fe—O键长越长,S—O键长越短,发生过渡态越晚,反应越难发生。通过反应过程中对铁原子自旋密度的分析可知,反应过程是两电子转移的过程,发生直接的氧转移反应。本文从理论上探讨了4价非血红素铁模型配合物磺化氧化反应的反应机理,为进一步研究金属离子效应加速磺化氧化反应提供了理论基础。
参考文献:
[1]QUEL,HORYN.Dioxygenactivationbyenzymeswithmononuclearnon-hemeironactivesites[J].ChemicalReviews, 1996, 96(7): 2607-2624.
[2]COSTASM,MEHNMP,JENSENMP,etal.Dioxygenactivationatmononuclearnonhemeironactivesites:enzymes,models,andintermediates[J].ChemicalReviews, 2004, 104(2): 939-986.
[3]ROHDEJU,INJH,LIMMH,etal.CrystallographicandspectroscopiccharacterizationofanonhemeFe(Ⅳ)=Ocomplex[J].Science, 2003, 299: 1037-1039.
[4]ROHDEJU,TORELLIS,SHANXP,etal.Structuralinsightsintononhemealkylperoxoiron(Ⅲ)andoxoiron(Ⅳ)intermediatesbyX-rayabsorptionspectroscopy[J].JournaloftheAmericanChemicalSociety, 2004, 126(51): 16750-16761.

图3B2//B1计算水平下[FeⅣ(O)(N4Py)]2+与苯硫基甲烷及其对位衍生物的反应能垒和反应过渡态的几何结构
Fig.3B2//B1 energy profiles for the reaction of [FeⅣ(O)(N4Py)]2+with thioanisole and its derivations and geometric details of the O-transfer transition states
[5]PARKJ,MORIMOTOY,LEEYM,etal.Metalioneffectontheswitchofmechanismfromdirectoxygentransfertometalion-coupledelectrontransferinthesulfoxidationofthioanisolesbyanon-hemeiron(Ⅳ)-oxocomplex[J].JournaloftheAmericanChemicalSociety, 2011, 133(14): 5236-5239.
[6]BISWASAN,PURIM,MEIERKK,etal.ModelingTauD-J:ahigh-spinnonhemeoxoiron(Ⅳ)complexwithhighreactivitytowardC-Hbonds[J].JournaloftheAmericanChemicalSociety, 2015, 137(7): 2428-2431.
[7]LIMMH,ROHDEJU,STUBNAA,etal.AnFeⅣ=Ocomplexofatetradentatetripodalnonhemeligand[J].ProceedingoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica, 2003, 100(7): 3665-3670.
[8]KAIZERJ,KLINKEREJ,OHNY,etal.NonhemeFeⅣOcomplexesthatcanoxidizetheC—Hbondsofcyclohexaneatroomtemperature[J].JournaloftheAmericanChemicalSociety, 2004, 126(2): 472-473.
[9]KLINKEREJ,KAIZERJ,BRENNESSELWW,etal.Structuresofnonhemeoxoiron(Ⅳ)complexesfromX-raycrystallography,NMRspectroscopy,andDFTcalculations[J].AngewandteChemieInternationalEdition, 2005, 44(24): 3690-3694.
[10]SASTRICV,OHK,LEEYJ,etal.Oxygen-atomtransferbetweenmononuclearnonhemeiron(Ⅳ)-oxoandiron(Ⅱ)complexes[J].AngewandteChemieInternationalEdition, 2006, 45(24): 3992-3995.
[11]KREBSC,FUJIMORIDG,WALSHC,etal.Non-hemeFe(Ⅳ)-oxointermediates[J].AccountsofChemicalResearch, 2007, 40(7): 484-492.
[12]QUEL.Theroadtonon-hemeoxoferrylsandbeyond[J].AccountsofChemicalResearch, 2007, 40(7): 493-500.
[13]MCDONALDAR,QUEL.High-valentnonhemeiron-oxocomplexes:synthesis,structure,andspectroscopy[J].CoordinationChemistryReviews, 2013, 257(2): 414-428.
[14]NEESEF.Theoreticalspectroscopyofmodel-nonheme[Fe(Ⅳ)OL5]2+complexesintheirlowesttripletandquintetstatesusingmultireferenceabinitioanddensityfunctionaltheorymethods[J].JournalofInorganicBiochemistry, 2006, 100(4): 716-726.
[15]SHAIKS,HIRAOH,KUMARD.Reactivityofhigh-valentiron-oxospeciesinenzymesandsyntheticreagents:ataleofmanystates[J].AccountsofChemicalResearch, 2007, 40(7): 532-542.
[16]KUMARD,HIRAOH,QUEL,etal.TheoreticalinvestigationofC—Hhydroxylationby(N4Py)FeⅣ=O2+:anoxidantmorepowerfulthanP450[J].JournaloftheAmericanChemicalSociety, 2005, 127(22): 8026-8027.
[17]GOTOY,MATSUIT,OZAKISI,etal.Mechanismsofsulfoxidationcatalyzedbyhigh-valentintermediatesofhemeenzymes:electron-transfervsoxygen-transfermechanism[J].JournaloftheAmericanChemicalSociety, 1999, 121(41): 9497-9502.
[18]SHAIKS,WANGY,CHENH,etal.ValencebondmodellinganddensityfunctionaltheorycalculationsofreactivityandmechanismofcytochromeP450enzymes:thioethersulfoxidation[J].FaradayDiscussions, 2010, 145: 49-70.
[19]PORROCS,SUTCLIFFEMJ,DEVISSERSP,etal.Quantummechanics/molecularmechanicsstudiesonthesulfoxidationofdimethylsulfidebycompoundⅠandcompound0ofcytochromeP450:whichisthebetteroxidant[J].TheJournalofPhysicalChemistryA, 2009, 113(43): 11635-11642.
[20]KHENKINAM,LEITUSG,NEUMANNR.Electrontransfer-oxygentransferoxygenationofsulfidescatalyzedbytheH5PV2Mo10O40polyoxometalate[J].JournaloftheAmericanChemicalSociety, 2010, 132(33), 11446-11448.
[21]BECKEAD.Density-functionalthermochemistry. Ⅰ.Theeffectoftheexchange-onlygradientcorrection[J].TheJournalofChemicalPhysics, 1992, 96(3): 2155-2160.
[22]BECKEAD.Density-functionalthermochemistry. Ⅲ.Theroleofexactexchange[J].TheJournalofChemicalPhysics, 1993, 98(7): 5648-5652.
[23]LEEC,YANGW,PARRRG.DevelopmentoftheColle-Salvetticorrelation-energyformulaintoafunctionaloftheelectrondensity[J].PhysicalReviewB, 1988, 37(2): 785-789.
[24]TOMASIJ,MENNUCCIB,CAMMIR.Quantummechanicalcontinuumsolvationmodels[J].ChemicalReviews, 2005, 105(8): 2999-3094.
[25]HAYPJ,WADTWR.Abinitioeffectivecorepotentialsformolecularcalculations.PotentialsforKtoAuincludingtheoutermostcoreorbitals[J].TheJournalofChemicalPhysics, 1985, 82(1): 299-310.
[26]FRIESNERRA,MURPHYRB,BEACHYMD,etal.Correlatedabinitioelectronicstructurecalculationsforlargemolecules[J].TheJournalofPhysicalChemistryA, 1999, 103(13): 1913-1928.
[27]WEIGENDF,AHLRICHSR.Balancedbasissetsofsplitvalence,triplezetavalenceandquadruplezetavalencequalityforHtoRn:designandassessmentofaccuracy[J].PhysicalChemistryChemicalPhysics, 2005, 7(18): 3297-3305.
[28]SCHAEFERA,HORNH,AHLRICHSR.FullyoptimizedcontractedGaussianbasissetsforatomsLitoKr[J].TheJournalofChemicalPhysics, 1992, 97(4): 257-2577.
[29]SCHAEFERA,HUBERC,AHLRICHSR.FullyoptimizedcontractedGaussianbasissetsoftriplezetavalencequalityforatomsLitoKr[J].TheJournalofChemicalPhysics, 1994, 100(8): 5829-5835.
Mechanismofsulfoxidationreactionbynon-hemeiron(Ⅳ)-oxo[FeⅣ(O)(N4Py)]2+complex
LIUYuan1,WANGYi1,LIUWenhua2,TANGZhe1,ZHANGAijie3
( 1.SchoolofBiologicalEngineering,DalianPolytechnicUniversity,Dalian116034,China;2.SchoolofMechanicalEngineeringandAutomation,DalianPolytechnicUniversity,Dalian116034,China;3.SchoolofChemistryandMaterialScience,ShanxiNormalUniversity,Linfen041001,China)
Abstract:The sulfoxidation of non-heme iron(Ⅳ)-oxo [FeⅣ(O)(N4Py)]2+complex were studied by density functional theory calculations. When thioanisole and its para derivatives were selected as substrate, the Mulliken charges of sulfur atom were increased with the electron donation power of para-substituted. The mechanism of sulfoxidation reaction was two-electron transfer reaction. The oxygen transfer reaction was two-state and its reactivity was determined by quintet energy barrier. The reaction energy barrier increased with the electron donation power of para-substituted, and the relative reactivity descended as follows: para-Me-thioanisole, thioanisole, para-Cl-thioanisole, para-CN-thioanisole, para-NO2-thioanisole.
Key words:non-heme; sulfoxidation reaction; density functional theory; oxygen transfer reaction
收稿日期:2015-10-27.
基金项目:辽宁省教育厅一般项目(L2013214);分子反应动力学国家重点实验室开放课题(K201511).
作者简介:刘 媛(1991-),女,硕士研究生;通信作者:王 一(1981-),女,讲师.
中图分类号:O641
文献标志码:A
文章编号:1674-1404(2016)03-0212-05
