
一个新的七元环吡咯-吡啶氢键分子激发态分子内质子转移的理论研究
2016-07-05 08:10袁慧娟郭续更
化学研究 2016年3期
关键词:氢键
袁慧娟,郭续更
(河南大学 化学化工学院,河南 开封 475004)
一个新的七元环吡咯-吡啶氢键分子激发态分子内质子转移的理论研究
袁慧娟,郭续更*
(河南大学 化学化工学院,河南 开封 475004)
摘要:运用含时密度泛函理论(TD-DFT)方法和以环己烷为溶剂的可极化连续模型(PCM),研究了2-[2-(1H-pyrrol-2-yl)-cyclopent-1-enyl]-pyridine(7-HB)发生激发态分子内质子转移(ESIPT)的反应机制.结果表明,7-HB分子被光激发到Franck-Condon区域后,在第一光学亮态(S1)上会发生一个超快的从Normal (N) 式到Tautomer (T)式的质子转移反应,其反应的能垒仅有0.05 eV.此外,在相同计算水平下,还研究了7-HB分子的吸收和发射光谱,所得结果与实验数据吻合得很好.
关键词:含时密度泛函理论;激发态分子内质子转移;七元环分子;氢键
近年来,激发态分子内质子转移(ESIPT)反应在荧光探针、光学材料等光电子领域[1-4]都有着广泛的应用,一直是研究者关注的焦点.众所周知的是,ESIPT分子都拥有质子给体(羟基或氨基等)和质子受体(羰基氧原子或吡咯氮原子等),可以形成一个或多个分子内氢键,这些分子内氢键能够与相邻的芳香环形成五元或六元氢键网络[5].在紫外光的照射下,ESIPT分子被激发到Franck-Condon区域的光学亮态后,一个或多个质子能够从质子给体转移到邻近的质子受体上,完成从Normal(N式)到Tautomer(T式)结构的转变.在大多数情况下,ESIPT反应都是超快的(飞秒数量级),并伴随着荧光的产生[5-8]. ESIPT分子的一个重要性质就是它的发射光谱和吸收光谱之间会出现较大的斯托克斯位移.
羟基型的ESIPT反应通常都是放热的,其反应速率都很快.但是,ESIPT很少发生在氨基型氢键体系上,因为氨基(N-H)质子的酸性比羟基(O-H)质子的酸性弱了很多.为了提高氨基型氢键体系发生ESIPT的能力,实验和理论学家进行了大量有益的尝试[9-12].一种行之有效的办法是在N-H质子上引入吸电子基团,增加N-H质子的酸性.最近,CHOU课题组[12]还发现了另一种提高氨基型氢键体系的ESIPT能力的方法.他们合成了两个分别拥有六元和七元环的吡咯-吡啶氢键分子(见图1),发现7-HB分子的ESIPT反应速率小于150 fs,比6-HB分子的1.4 ps的ESIPT速率快了很多.此外,他们还采用TD-CAM-B3LYP/6-31+G(d,p)方法研究了6-HB的第一激发态(S1)的势能面,发现6-HB在ESIPT过程中需要克服2.3 kcal·mol-1的能垒[12].由于飞秒时间分辨荧光光谱测得的7-HB的ESIPT速率很快,他们推测7-HB的ESIPT反应应该是一个无能垒的过程[12].为了研究7-HB分子的超快ESIPT反应机理,我们采用密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)方法计算了7-HB分子的沿质子转移方向的势能曲线,还研究了它的基态(S0)和第一激发态(S1)的稳定构型,以及吸收和发射光谱.大量研究表明,DFT和TD-DFT方法能够很好地处理一些有机共轭分子的结构和光谱性质[13-14].
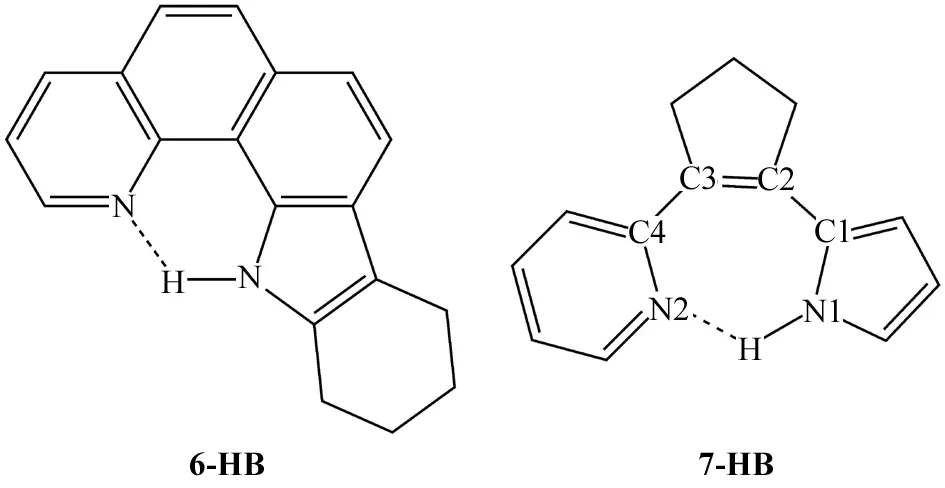
图1 6-HB和7-HB分子的结构Fig.1 Structures of 6-HB and 7-HB molecules
1计算方法
运用DFT方法[15],优化了7-HB分子的N式的基态(S0)平衡构型. 采用TD-DFT方法[16-17],优化了7-HB分子的N式和T式的第一激发态(S1)的结构. 选取的密度泛函是CAM-B3LYP[18],基组是6-31+G(d,p)[19]. 在相同水平下还进行了频率计算,所有频率都是正值,没有虚频存在,表明在这两种水平下优化得到的结构都是稳定构型. 此外,在TD-CAM-B3LYP/6-31+G(d,p)的计算水平下,还研究了7-HB分子的吸收和发射光谱. 为了探索质子转移过程的详细机制,我们分别运用CAM-B3LYP和TD-CAM-B3LYP方法,将沿质子转移的路径方向的N1-H的距离固定为不同的值,其他参数不加任何限制,优化得到了7-HB分子在S0和S1的势能面上的能量变化曲线(CEPs). 为了考察溶剂对7-HB分子的结构和光谱性质的影响,以上所有的计算都采用了以环己烷为溶剂的极化连续模型(PCM)[20]. 所有的DFT和TD-DFT计算都是在Gaussian 09程序下完成的[21].
2结果与讨论
2.1几何构型
图2展示了7-HB分子的N式的S0结构(N-S0)以及N式和T式的S1结构(N-S1和T-S1). 从图2可以看出,在7-HB分子中,N1-H 官能团与邻近的N2原子能形成一个分子内氢键,即N1-H…N2,它能够与周围的4个碳原子(C1、C2、C3和C4)形成一个七元环结构. 在N-S0结构中,N1-H…N2氢键键长是0.179 nm. 但是,在N-S1结构中,N1-H…N2氢键键长变成了0.162 nm,比N-S0的氢键键长缩短了0.017 nm. 一般来说,氢键键长越短,氢键强度越大,就越有利于后续的ESIPT反应.在发生 ESIPT反应以后,在T-S1结构中的N1…H-N2氢键键长进一步缩短到0.159 nm.
表1列出了7-HB分子的N-S0、N-S1和T-S1结构的相对能量和一些重要的结构参数. 从表1可以看出,N-S0的能量最低,表明7-HB分子的N-S0结构是一个全局极小点. 在N-S0结构中,N1和N2原子间的距离RN1-N2是0.272 nm,DN1-C1-C2-C3和DN2-C4-C3-C2二面角分别是7.4°和14.3°. N-S1结构的能量比N-S0的能量高了3.17 eV. 与N-S0的结构参数相比,N-S1的RN1-N2距离缩短了0.010 nm,DN1-C1-C2-C3和DN2-C4-C3-C2二面角分别降低了2.7°和7.3°,增加了7-HB分子结构中七元环的共面程度,有利于ESIPT反应的发生. 另一个推动ESIPT过程的因素是T-S1结构的能量比N-S1的能量略低,有利于从N-S1到T-S1结构的转变.
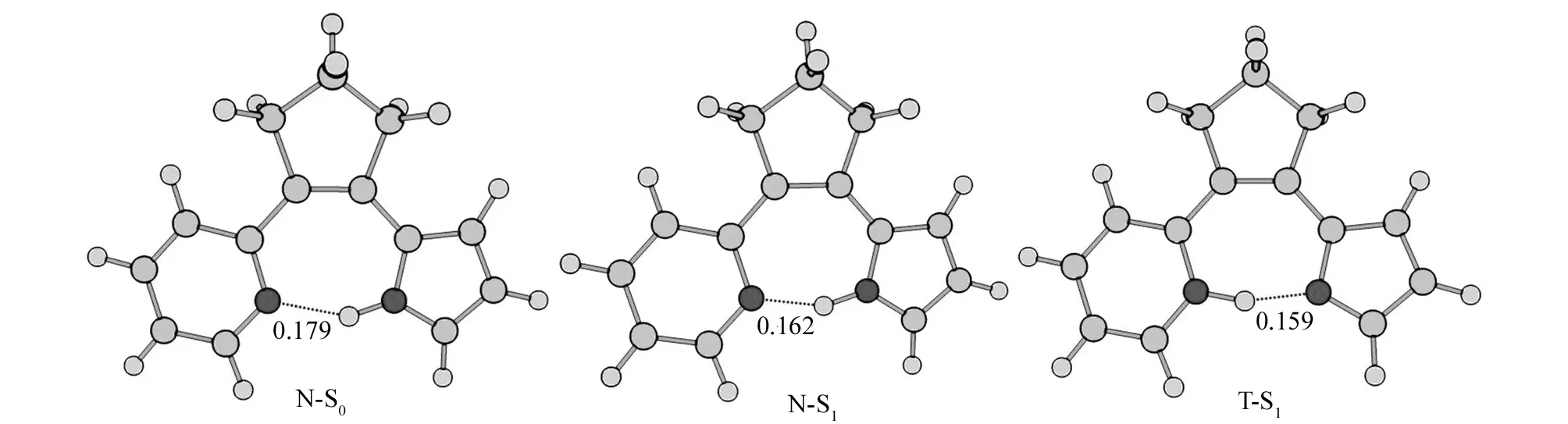
图2 用CAM-B3LYP/PCM 和TD-CAM-B3LYP/PCM方法优化得到的7-HB分子N式的S0结构以及N式和T式的S1结构(单位:nm)Fig.2 CAM-B3LYP/PCM and TD-CAM-B3LYP/PCM optimized the S0 structure in the N form and the S1 structures in the N and T forms in 7-HB molecule (distance in nm)

表1 7-HB分子的三种结构的相对能量(单位:eV)和一些重要的结构参数(键长:nm;二面角:°)
2.2吸收和发射光谱
表2列出了由TD-CAM-B3LYP/6-31+G(d,p)/PCM方法计算得到的7-HB分子的垂直激发能和垂直发射能,以及可用的实验数据. 从表2可以看出,7-HB的垂直激发能的计算值是3.60 eV,相应的振子强度是0.610,与实验值(3.28 eV[12])相符合,表明在实验上观测到的吸收峰应该源于7-HB的N式结构的贡献. N-S1和T-S1的垂直发射能分别是3.02和2.61 eV. 显然,在实验上观测到的发射峰(2.06 eV)应该源于7-HB的T式结构的发射. 从表2也可以看出,7-HB的垂直激发能是来自于最高占据分子轨道(HOMO)到最低空分子轨道(LUMO)的电子跃迁.图3展示了7-HB分子的HOMO和LUMO轨道轮廓图. 从图3可以明显看出,HOMO轨道的电子主要分布在吡咯环上,而LUMO轨道的电子主要分布在吡啶环上,表明7-HB分子从HOMO到LUMO的电子跃迁具有明显的电荷转移的特征. 一般来说,CAM-B3LYP密度泛函能够很好地处理具有电荷转移特征的分子体系[22],因此我们在7-HB分子的理论计算中采用了这种方法. 图4展示的是由TD-CAM-B3LYP/6-31+G(d,p)/PCM方法模拟得到的7-HB分子的吸收和发射光谱. 从图4可以看出,7-HB的T-S1的发射光谱与N-S0的吸收光谱相比发生了明显的斯托克斯红移,这满足一个分子发生ESIPT反应的基本特征.
2.3质子转移势能面
图5是计算得到的7-HB分子的S1态和S0态的沿质子转移方向的势能曲线(CEPs)图. 在图5中我们以CAM-B3LYP/PCM和TD-CAM-B3LYP/PCM方法分别优化得到的N-S0和N-S1的构型作为CEPs的起点,而其他的点则通过固定N1-H的距离在相同的计算水平下进行优化得到,用各个点的能量值绘制成CEPs图.
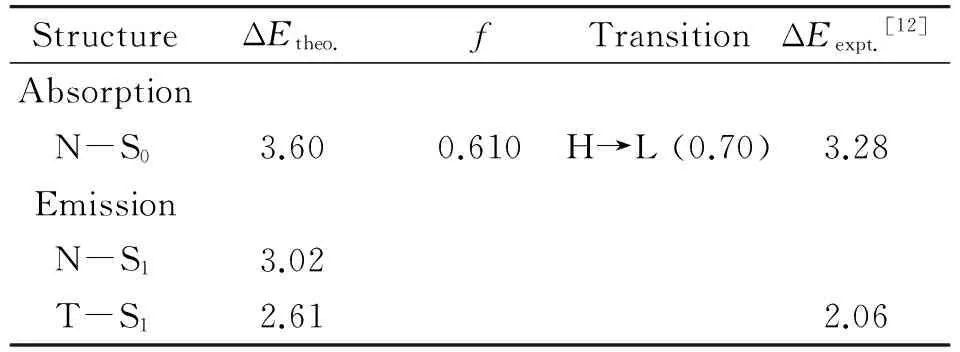
表2 用TD-CAM-B3LYP/PCM 方法计算的7-HB的

图3 7-HB分子的HOMO和LUMO轨道轮廓图Fig.3 HOMO and LUMO molecular orbitals of the 7-HB molecule
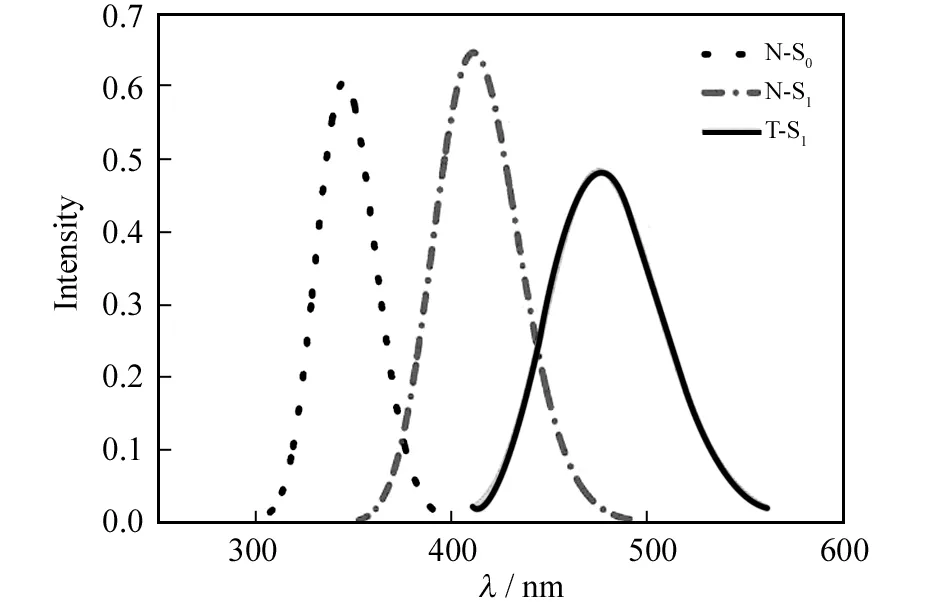
图4 7-HB分子的吸收与发射光谱Fig.4 Simulated absorption and emission spectra for 7-HB molecule
观察7-HB分子的势能面变化曲线图可以看出,在S1态的质子转移势能面变化曲线上有一个最高点明显存在,这表明7-HB分子在由N-S1到T-S1的质子转移过程中需要克服一个很小的能垒,其能垒值为0.05 eV,比在6-HB分子的S1态的质子转移势能面上的能垒(0.1 eV[12])略低,使得7-HB分子比6-HB分子的ESIPT过程要快,这些分析与实验发现是一致的[12]. 如前所述,7-HB分子经过ESIPT反应生成T-S1后,会以发出荧光的形式辐射失活到T-S0态. 由于T-S0态的结构很不稳定,它会迅速地经过反向的基态质子转移(RGSHT)反应弛豫回到N-S0态. 从图5可以明显看出,7-HB分子在由T-S0到N-S0的RGSHT反应是一个无能垒的过程.

图5 7-HB分子沿质子转移方向的势能曲线Fig.5 Constrained energy profiles (CEPs) of 7-HB along the proton transfer coordinate
3结论
采用CAM-B3LYP/PCM方法,优化得到了7-HB分子的N-S0的平衡构型. 运用TD-CAM-B3LYP/PCM方法,优化得到了7-HB分子的N-S1和T-S1的平衡构型. 在TD-CAM-B3LYP/PCM水平下,计算得到了7-HB分子的吸收和发射光谱. 计算表明,在7-HB中,N-S0结构是一个全局极小点,而T-S1比N-S1结构的能量略低,这有利于后续的ESIPT反应. 在N-S0构型下,用TD-CAM-B3LYP/PCM方法计算得到的7-HB分子的吸收峰位于3.60 eV,与实验值(3.28 eV)相符合. 分别在N-S1和T-S1的构型下,用相同方法计算得到的7-HB分子的发射峰分别位于3.02和2.61 eV,证实了在实验上观测到的发射峰(2.06 eV)应该源于T-S1构型的发射. 从CEPs势能面曲线上可以看出,当7-HB分子被光激发到S1亮态后,它从N-S1到T-S1的质子转移应该是一个超快的过程,其反应能垒仅有0.05 eV,而由T-S0到N-S0的质子转移是一个无能垒的过程.
参考文献:
[1] LANDGE S M, TKATCHOUK E, BENTEZ D, et al. Isomerization mechanism in hydrazine-based rotary switches: Iateral shift, rotation, or tautomerization? [J]. J Am Chem Soc, 2011, 133(25): 9812-9823.
[2] KWON J E, PARK S Y. Advanced organic optoelectronic materials: harnessing exci-ted-state intramolercular proton transfer (ESIPT) process [J]. Adv Mater, 2011, 23(32): 3615-3642.
[3] ZHAO J Z, JI S M, CHEN Y H, et al. Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials [J]. Phys Chem Chem Phys, 2012, 14(25): 8803-8817.
[4] YAO D, ZHAO S, GUO J, et al. Hydroxyphenyl-benzothiazole based full color organic emitting materials generated by facile molecular modification [J]. J Mater Chem, 2011, 21(11): 3568-3570.
[5] HOUARI Y, CHIBANI S, JACQUEMIN D, et al. TD-DFT assessment of the excited state intramolecular proton transfer in hydroxyphenylbenzimidazole (HBI) dyes [J]. J Phys Chem B, 2015, 119(6): 2180-2192.
[7] EI-SAYED Y S, El-Daly S A, GABER M. Spectral behavior and laser activity of 3-(4′-dimethylaminophenyl)-1-(1H-phrrol-2-yl) prop-2-en-1-one (DMAPr P) [J]. Opt Laser Technol, 2010, 42(2): 397-402.
[8] CHEN K Y, CHENG Y M, LAI C M, et al. Ortho green fluorescence protein synthetic chromophore;excited-state intramolecular proton transfer via a seven-membered-ring hydrogen-bonding system [J]. J Am Chem Soc, 2007, 129(15): 4534-4535.
[9] TSENG H W, LIU J Q, CHEN Y A, et al. Harnessing excited-state intramolecular proton-transfer reaction via a series of amino-type hydrogen-bonding molecules [J]. J Phys Chem Lett, 2015, 6(8): 1477-1486.
[10] CHEN C L, TSENG H W, CHEN Y A, et al. Insight into the amino-type excited-state intramolecular proton transfer cycle using N-tosyl derivatives of 2-(2′-aminophenyl) benzothiazole [J]. J Phys Chem A, 2016, 120(7): 1020-1028.
[11] TSENG H W, LIN T C, CHEN C L, et al. A new class of N-H proton transfer molecules: wide tautomer emission tuning from 590 nm to 770 nm via a facile, single site amino derivatization in 10-aminobenzo[h]qui-nolone [J]. Chem Commun, 2015, 51(89): 16099-16102.
[12] ZHANG Z, HSU Y H, CHEN Y A, et al. New six-and seven-membered ring pyrrole-pyridine hydrogen bond systems undergoing excited-state intramolecular proton transfer [J]. Chem Commun, 2014, 50(95): 15026-15029.
[13] 李洁琼, 赵清岚. 三种席夫碱-Ni(II)配合物的电子结构和吸收光谱的理论计算[J]. 化学研究, 2014, 25(5): 497-503.
[14] 李云飞, 王新收, 吴文鹏. 一种基于苯并噻二唑衍生物的F-荧光探针分子的理论研究[J]. 化学研究, 2015, 26(6): 575-578.
[15] GROSS E U K, KOHN W. Local density-functional theory of frequency-dependent linear response [J]. Phys Rev Lett, 1985, 55(26): 2850.
[16] RUNGE E, GROSS E U K. Density-functional theory for time-dependent systems [J]. Phys Rev Lett, 1984, 52(12): 997.
[17] FURCHE F, AHLRICHS R. Adiabatic time-dependent density functional methods for excited state properties [J]. J Chem Phys, 2002, 117(16): 7433-7447.
[18] YANAI T, TEW D P, HANDY N C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP) [J]. Chem Phys Lett, 2004, 393(1): 51-57.
[19] HEHRE W J, DITCHFIELD R, POPLE J A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules [J]. J Chem Phys, 1972, 56(5): 2257-2261.
[20] CANCES E, MENNUCCI B, TOMASI J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics [J]. J Chem Phys, 1997, 107(8): 3032-3041.
[21] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09 [CP]. Revision A. 02, Wallingford CT: Gaussian, Inc., 2009.
[22] GUO X G, CAO Z X. Low-lying electronic states and their nonradiative deactivation of thieno[3,4-b]pyrazine: an ab initio study [J]. J Chem Phys, 2012, 137(22): 224-313.
[责任编辑:吴文鹏]
收稿日期:2016-04-15.
基金项目:国家自然科学基金项目(21503069).
作者简介:袁慧娟 (1990-),女,硕士生.研究方向:理论与计算化学.*通讯作者,E-mail:xgguo@henu.edu.cn.
中图分类号:O641.3
文献标志码:A
文章编号:1008-1011(2016)03-0294-05
Theoretical study on excited-state intramolecular proton transfer of a new seven-membered ring pyrrole-pyridine bond molecule containing hydrogen bonding
YUAN Huijuan, GUO Xugeng*
(CollegeofChemistryandChemicalEngineering,HenanUniversity,Kaifeng475004,Henan,China)
Abstract:Time-dependent density functional theory (TDDFT) approaching along with the polarizable continuum model (PCM) using cyclohexane as a solvent was used to explore the exci-ted-state intramolecular proton transfer (ESIPT) reaction of 2-[2-(1H-pyrrol-2-yl)-cyclopent-1-enyl]-pyridine (7-HB). The result indicated that while the 7-HB molecule was photoexcited into the Franck-Condon region, it could undergo an ultrafast proton transfer reaction from the normal (N) to tautomer (T) forms in the optically bright first excited singlet state (S1), with a negligible barrier of 0.05 eV. In addition, the absorption and emission spectra of 7-HB were also calculated at the same theoretical level. It was found that our theoretical predictions were in good agreement with the available experimental findings.
Keywords:TD-DFT; excited-state intramolecular proton transfer; seven-membered ring molecule; hydrogen bonding
猜你喜欢
高中数理化(2022年14期)2022-08-15
波谱学杂志(2021年3期)2021-09-07
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
杭州化工(2020年2期)2020-01-16
沈阳师范大学学报(自然科学版)(2019年2期)2019-06-19
原子与分子物理学报(2019年5期)2019-04-28
信阳师范学院学报(自然科学版)(2018年1期)2018-08-09
天津师范大学学报(自然科学版)(2016年4期)2016-12-14
中学化学(2015年12期)2016-01-19
原子与分子物理学报(2015年2期)2015-03-23
