
一种基于宏基因组模拟数据的生物标志物筛选方法
2016-07-23 01:38王晓君滕琳
江苏农业科学 2016年5期
关键词:生物标志物
王晓君++滕琳

摘要:鉴于生物圈中微生物资源的巨大开发潜力以及测序技术不断发展,宏基因组学研究的不断深入,微生物群落已经被看作一个整体来进行分析并且已经得到广泛应用。然而由于微生物的多样性以及微生物菌群的复杂性,使得精确确定和定量宏基因组数据中的分类单元成为宏基因组数据分析的难点。已有的宏基因组数据标记分析工具无法解决微生物群落预测结果重现的稳健性、准确性以及处理非冗余标记物方面遇到的问题。笔者提出了一个新的基于宏基因组自助抽样(metagenomic bootstrap)的生物标志物选择方法,它结合了mRMR(minimal redundancy maximal relevance)和自助抽样方法(bootstrapping),可以更加稳健、准确而有效地通过对宏基因组数据的挖掘实现非冗余标记物的筛选。基于模拟数据集,通过其与2种自上而下的方法(Metastats、LEfSe)以及自下而上的方法(Wilcoxon秩和检验)进行对比,表明本方法可以在较高准确率的基础上更加稳健地选择更多的非冗余生物标志物。
关键词:宏基因组;生物标志物;mRMR;自助抽样法
中图分类号: Q789文献标志码: A文章编号:1002-1302(2016)05-0056-04
微生物一直被人们视为巨大的生物资源,尤其是其庞大的基因组数据包含有大量不为人知的新功能基因,将对人类的生产、生活做出卓越贡献[1]。然而,微生物资源中九成以上的微生物是不可培养的,也就意味着在新基因探索的道路上,人类面临着不小的困难。新一代测序技术的出现将帮助人们揭示不可(或难)培养微生物的基因组信息,从而发现新的微生物或新的功能基因。随着微生物基因组数据库的不断壮大,人们普遍意识到宏基因组数据分析的难点,宏基因组数据中生物标志物的鉴定以及应用非常重要。但宏基因组数据分析并不简单,研究显示,微生物群落展现出了非同一般的主体间可变性,更不可思议的是,此可变性竟然出现在人类和环境菌群中[2-3]。目前,人们已知的宏基因组生物标志物的鉴定方法有2种:一种是自下而上的方法,主要包括Wilcoxon秩和检验[4],测试每个分类单元,选择群体间具有差异的元素作为标志物;另外一种是自上而下的方法,主要包括Metastats、LEfSe。虽然这2种方法都可以用来统计评估宏基因组数据的差异,对生物标志物进行鉴定,但这些方法很难解决数据分析结果重现的稳健性、冗余性等问题。笔者提出一个自上而下的结合mRMR[5]和自助抽样法从微生物宏基因组样本中筛选生物标志物的方法,此方法首先分析微生物群落的整体分布,然后进行生物标志物筛选,不同于传统生物标志物筛选的是,它结合了mRMR,能更为有效地避免了生物冗余标志物这一难题。
1材料与方法
1.1模拟数据集的产生
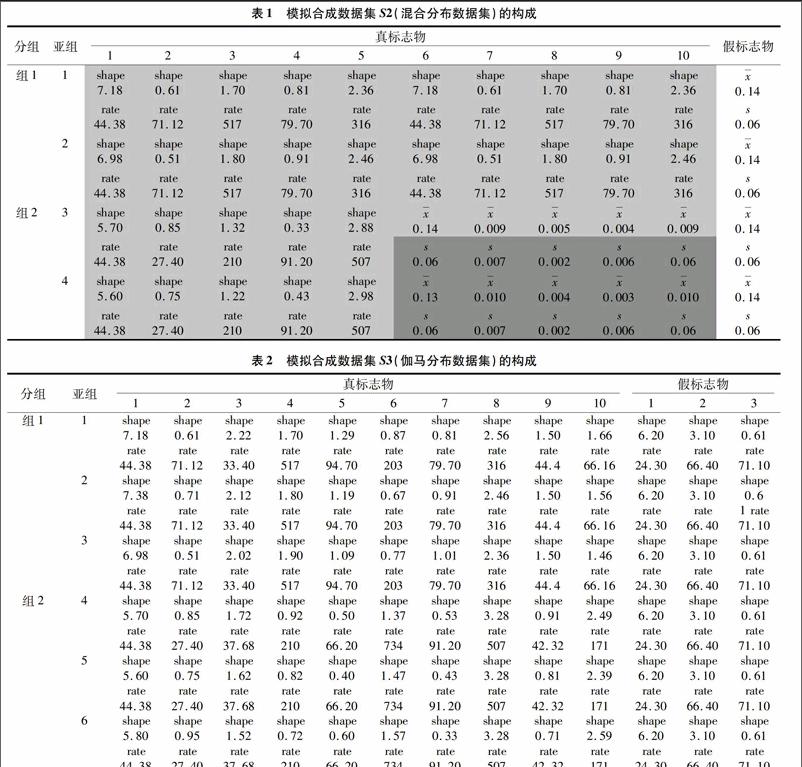
S1模拟数据集:根据文献,微生物群落的分类分布都遵循正态分布,故而基于正态分布,产生模拟数据集S1(S1未列出,仅说明特性,其具体的结构类似于下面即将产生的数据集S3,只是在生成数据时产生的是正态分布的数据,不同分类之间的差异指的是均值差异。)。S1中共有1 000个变量和120个样本,包含2个分类(每个分类包含3个亚类,每个亚类包含20个样本)。对于每一个样本来说,都包含10个真标志物组(10个变量/组)和1个假标志物组(900个变量/假标志物)。数据集S1的特性是真标志物中的2个分类组均值差异较大,在每个分类内部,亚类之间的差异很小(在每个标志物组内,虽然生成数据时没有差异,但是由于随机函数的缘故,差异在所难免)。S2模拟数据集:笔者分析以前本实验室口腔微生物宏基因组数据[6]发现,微生物群落的宏基因组数据的分布不单是正态分布这么简单,往往会有10%的变量符合正态分布和伽玛分布2种混合分布模式,因此基于正态和伽玛混合分布产生模拟数据集S2(表1)。数据集S2有2个重要特性:第一,对于真标志物,2个分类组参数shape(伽玛分布中的1个重要参数)或者均值差异较大,每个分类内部亚类之间的差异较小;第二,对于假标志物,它们在分类、亚类之间均值没有差异(每个标志物组内随机差异如S1所述)。此外,处在相同标志物组内的变量被认为是冗余的变量。S3模拟数据集:根据之前口腔样本数据发现,超过40%的变量仅符合伽玛分布,因此基于伽玛分布产生模拟数据集S3(表2)。数据集S3区别于S2的特性在于真标志物中S3数据集的2个分类组在参数shape上差异较大,在每个分类内,亚类之间的差异较小。
在真标志物中,一个小方格是一个25(样本)×10(变量)的矩阵。矩阵每一列的值都是由正态分布函数或者伽马分布函数(利用R语言中rnorm或者rgamma函数实现)产生的。表格中填充浅灰色的格子表示由伽马分布函数产生,填充深灰色的格子表示由正态分布函数产生。而假标志物组中,每一个都是一个25(样本)×900(变量)的矩阵,其数值由正态分布函数产生。
每个包含在真标志物中小方格都是一个20(样本)×10(变量)的矩阵。矩阵每列的值都由伽马分布函数(利用R语言中rgamma函数实现)产生。但对于假标志物组,每个格子都是一个20(样本)×300(变量)的矩阵,其数值也是由伽马分布函数产生。
1.2分析流程
归一化:为了减少原始数据的噪声,增强mRMR方法选择具有识别能力的变量,模拟数据集需要进行离散化,即用原始数据的均值(μ)和标准差(σ)对数据进行离散化。任何数据大于μ+σ/2转换为1,小于μ-σ/2转换为-1,其他数据转换为0。同时,原始的读长数目需要进行归一化,转换为相对丰度,即每个变量的读长数除以所有样本在该变量中的读长总数,每个变量的总和为1(变量中80%都是0将被忽略)。主要分析流程:归一化后的数据采用变量筛选和自助重抽样2个步骤进行去冗余,具体流程见图1。第一步的参数为1~M,其中M为第一次变量筛选时被mRMR筛选出的候选变量,用于区分不同样本(可能含有冗余变量);第二步为自助重抽样,参数为2~B;第三步为变量排序,参数是3~M′,这些变量是上一步抽样中被mRMR选出的,当所有的自助重抽样与变量选取完成后,按照变量出现次数进行排序,选取最终M′个变量作为最终用户需要的变量(M>M′)。
2结果与讨论
2.1基于宏基因组的自助抽样方法的参数选择
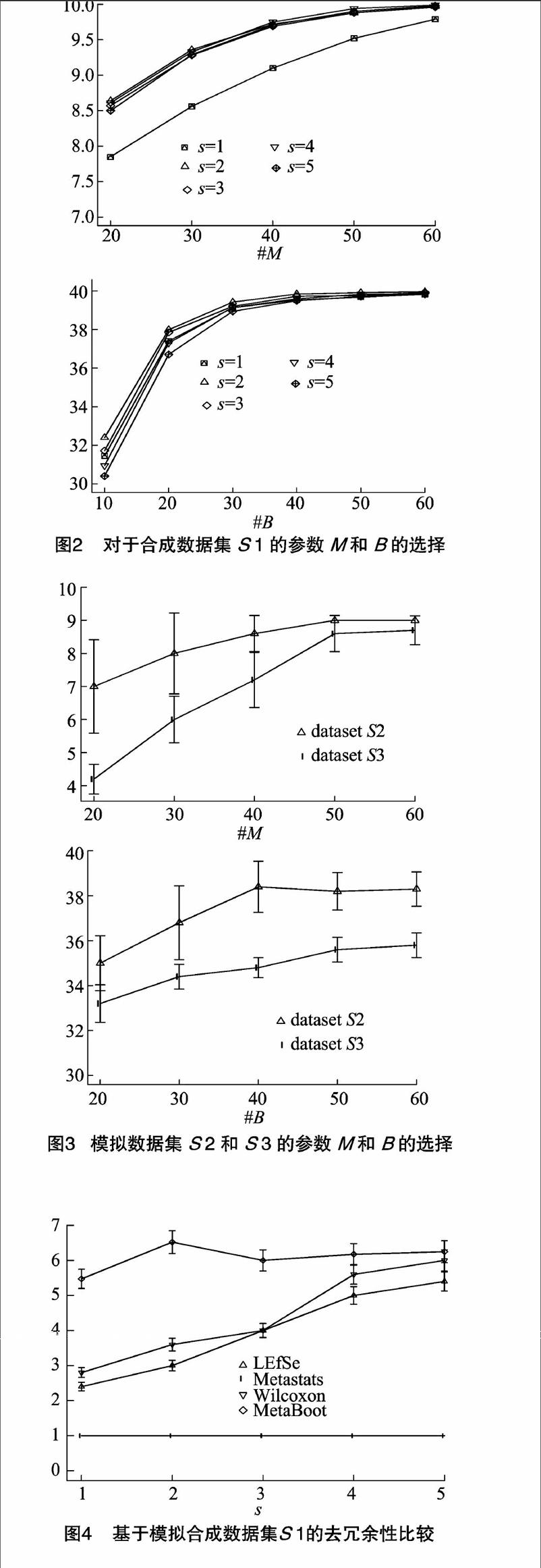
此方法过程主要包括3步:变量筛选步骤、自助重抽样和变量筛选过程以及变量排序,整个过程包含3个主要参数,分别是M、M′、B,它们对于选择生物标记物的质量有重大影响。对于模拟数据集S1来说,参数M设置为50。当M等于50时,几乎全部的非冗余变量都会被mRMR从1 000个变量中选出,考虑到计算的效率,50已经足够,因此没有选择更高的标准。对于参数B的选择,笔者设置了一系列自助重抽样次数的梯度,结果显示,当B超过40时,被选择出来的真标记物s不再增加(由于原始数据s的不固定性,因此选择多个s来表征数据的变化趋势)(图2)。B值设为40。同样的道理,对于数据集S2、S3中M′的选择,结果与S1具有一致性(图3)。由于S1只包含10个真标记物组,因此参数M′设置为10(最为理想的结果是每个标志物组中含有1个变量M′)。因此,将整体数据集参数M、B、M′分别设置为50、40、10。对本研究中基于自助抽样的生物标志物选择方法进行了去冗余性和准确性分析,来考察本方法是否更适合于宏基因组数据分析。冗余率、非冗余率计算公式如下:
冗余率=冗余的标志物数目选择标志物总数目×100%;(1)
非冗余率=特异的真生物标记物数选择标志物总数目×100%。(2)
2.2去冗余性分析
由图4可知,对于数据集S2、S3,本研究的新方法得到了最好的分析结果(表3),同时在数据集S1中,也得到了很好的区分效果。此外,本研究基于自助抽样的新方法较其他方法得到了更多的非冗余真标志物。宏基因组数据量庞大,各种各样的微生物基因片段都包含其中,表征微生物种属特性及其功能的特异性标准是研究生物标志物的意义所在。在复杂的数据库中寻找特异的生物标志物来重构菌群的复杂性,因此其选择的冗余性不可避免。本试验基于自助抽样方法很好地解决了冗余性这个难题,对于后续宏基因组工作有重要的应用价值。
2.3稳健性分析
基于3个模拟数据集,笔者分析比较了本方法与其他已经在宏基因组研究中应用的方法(如LEfSe、Metastats、Wilcoxon)在稳健性方面存在的差异。对于每种方法,选择100个生
物标志物(等于每个数据集中真生物标志物数目)计算100个生物标志物的百分率,结果见图5、表4。在已有的研究方法中,Wilcoxon在3个模拟数据集中的稳健性是最高的,本方法与Wilcoxon方法在3个数据集上相当,甚至表现更好。基于宏基因组数据生物标志物选择的方法,选择出的生物标志物具有较少的冗余固然重要,但是能够选择出在不同分组样本中有差异的生物标志物是前提。本方法的稳健性能够保证选出的生物标志物能够代表或者区分不同的样本,只有这样的生物标志物才有生物学意义。
2.4分类准确性分析
分类准确性是生物标志物选择方法是否具有竞争力的重要指标。分类准确率计算公式如下:
分类准确率=准确分类的样本数目测试样本中样本总数×100%。(3)
此部分只采用S2及S3作为验证分类准确率与否的数据集,由于S1数据集内部区分非常明显,对于任何一种区分方法都能实现很好的分类结果,因此在后2个数据集中分析比较这几种方法的优劣更有意义。分类时,使用这4种方法选择的10个标志物来建模。其中,每个数据集都有2类,每类含有60个样本,采用50个样本作为训练数据集,10个样本作为检验数据集,结果显示,在2个数据集准确性的分析中,基于自助抽样的方法较其他3种方法具有更高的分类准确性以及最小的区分结果变异性,即最小的s(标准方差)值(图6)。分类准确性是笔者选择方法的一个重要指标,基于自助抽样方法与其他生物标志物选择方法相比,在分类准确性方面具有非常明显的优势,在今后对于宏基因组研究中,本方法可以很好地实现对于生物标志物的选择。
3结论
目前宏基因组数据缺乏生物标志物的背景信息,使得利用各种方法预测宏基因组生物标志物变得困难[7]。笔者提出了将基于自助抽样的方法用于宏基因组生物标志物的鉴定,它是一个自上而下的方法,结合了mRMR方法和自助重抽样技术。基于模拟数据集,通过其与2种自上而下的方法(Metastats、LEfSe)以及自下而上的方法(Wilcoxon秩和检验)进行对比,表明本方法可以在较高准确率的基础上更加稳健地选择更多的非冗余生物标志物。但本方法在鉴定功能性的生物标志物方面不是非常理想,还需进一步完善。
参考文献:
[1]Ndimba B K,Ndimba R J,Johnson T S,et al.Biofuels as a sustainable energy source:An update of the applications of proteomics in bioenergy crops and algae[J]. Journal of Proteomics,2013,93:234-244.
[2]Pedros-Alio C. Marine microbial diversity:can it be determined? [J]. Trends in Microbiology,2006,14(6):257-263.
[3]Liao,L,Xu X W,Jiang X W,et al. Microbial diversity in deep-sea sediment from the cobalt-rich crust deposit region in the Pacific Ocean[J]. Microbiology Ecology,2011,78(3):565-585.
[4]Bauer D F. Constructing confidence sets using rank statistics[J]. Journal of the American Statistical Association,1972,67(339):687-690.
[5]Ding C,Peng H C. Minimum redundancy feature selection from microarray gene expression data[C]. Proceedings of the 2003 IEEE Bioinformatics Conference,2003:523-528.
[6]Huang S,Li R,Zeng X W,et al. Predictive modeling of gingivitis severity and susceptibility via oral microbiota[J]. The ISME Journal,2014,8(9):1768-1780.
[7]高岳. 应用宏基因组技术从微生物中获得活性物质的研究进展[J]. 江苏农业科学,2014,42(1):5-8.赵剑波,郭继英,姜全,等. 桃抗重茬砧木GF677组培快繁技术[J]. 江苏农业科学,2016,44(5):60-61,68.
猜你喜欢
云南中医中药杂志(2017年7期)2017-07-25
分析化学(2017年7期)2017-07-12
中国医学创新(2017年14期)2017-06-08
南水北调与水利科技(2017年1期)2017-02-27
