
PCR-based methodologies for detection and characterization of Listeria monocytogenes and Listeria ivanovii in foods and environmental sources
2017-05-22 09:01JinQingChenStephnieHeleyPtrickRegnPongpnLksnlmiZonglinHu
食品科学与人类健康(英文) 2017年2期
Jin-Qing Chen,Stephnie Heley,Ptrick Regn,Pongpn Lksnlmi,Zonglin Hu
a Winchester Engineering&Analytical Center(WEAC),HFR-NE460,Office of Regulatory Affairs,US Food and Drug Administration,109 Holton Street,Winchester,MA 01890,USA
b MD DHMH Laboratories Administration,Division of Microbiology,Room 528,1770 Ashland Ave.Baltimore,MD 21205,USA
Abstract Listeria monocytogenes is an important foodborne pathogen responsible for listeriosis,a fatal disease.It is widely distributed in various foods and environmental sources.In this review,we focused on addressing PCR-based technologies,including conventional PCR,qPCR and droplet digital PCR(ddPCR).Specificall,we described(a)conventional PCR and mono-,duplex-and multiplex-qPCR methodologies;(b)development and applications of gene HlyA-,Iap-,PrfA-and SsrA-based conventional and qPCR assays as well as PCR assays targeting newly identifie gene targets for specifidetection of L.monocytogenes;differentiation ofiviable from dead L.monocytogenes by qPCR in conjugation with propidium monoazide pretreatment;PCR-based serotype identificatio of L.monocytogenes isolates;PCR-based detection of L.ivanovii,infecting ruminants,differentiation of L.monocytogenes from other Listeria species;and sigB-gene based PCR identificatio of Listeria spp;(c)applications of ddPCR in detection of L.monocytogenes;and(d)application of qPCR assays in detection and subtyping of L.monocytogenes in milk and dairy products;meats,meat products and meat-processing environment;and seafood,seafood products and processing environment.Our goal was to provide a relatively comprehensive overview of PCR-based methodologies available in detection,characterization and subtyping ofivarious strains of L.monocytogenes in foods and environmental sources.
Keywords: Foodborne pathogens;Listeria monocytogenes;Listeria ivanovii;qPCR
1.Introduction
Listeria monocytogenes(L.monocytogenes),a gram-positive bacterium and a member of the genusListeria,is a ubiquitous and intracellular pathogen responsible for listeriosis,a fatal disease exhibiting the symptoms of abortion,neonatal death,septicemia,and meningitis.L.monocytogenesis found among pregnant women and their fetuses,immunocompromised persons,aging adults,patients with AIDS disease,cancer,organ transplant,and steroid treatment etc.with general mortality rate of 20%-30% [1-3].Owning to its capabilities of surviving in foods under adverse conditions and of growing at low refrigeration temperatures (such as 4°C),L.monocytogenesis widely distributed in the environment and various types of foods.The consumption ofL.monocytogenes-contaminated foods has been linked to major outbreaks in U.S.and Europe[4-7].A critical step toward control and prevention oflisteriosis outbreaks is to detectL.monocytogenesin environmental and food sources as rapidly as possible.A number of methodologies,including traditional culture-based methods and nucleic acid-based methods,have been developed and applied.In this review,we focused on description of the development and application ofivarious PCR-based methodologies,including conventional,real-time quantitative PCR(qPCR)and droplet digital PCR(ddPCR)and their applications in detection and characterization ofL.monocytogenesandL.ivanoviiin environmental and food sources.We also aim at providing a relatively complete summary ofivarious PCR technologies combined with other technologies that can be applied,in detection,characterization and subtyping ofL.monocytogenesandL.ivanoviiin food and environmental sources.
2.PCR-based methodologies
2.1.Conventional polymerase chain reaction(PCR)
Traditional culture-based methods in detection ofL.monocytogenesin various environmental settings and food matrices are labor-intensive and time-consuming.PCR-based methodologies have been applied in detection ofL.monocytogenes.
Conventional PCR is anin vitroenzymatic amplificatio of a targeted DNA sequence based upon oligonucleotide primerdirected DNA synthesis by a DNA polymerase.In PCR assay,two primers complementary to 3′ends of each of sense and anti-sense strand of target DNA are designed to flan the DNA sequence to be amplified Two primers are supplied in a buffered reaction mixture containing DNA polymerase plus four dNTPs,cofactors(e.g.Mg2+)and DNA template.Its general protocols consist of 3 steps that are temperature-controlled:(i)denaturation of double stranded DNA template into single stranded DNA at 95°C;(ii) hybridization (annealing) of two primers to their complementary regions of DNA template at different temperatures set according to melting temperatures(Tm)of two primers for 30-60s;which is typically set at about 3-5°C below Tm of the primers used;and(iii)extension(synthesis)of DNA from the sites dictated by primers at 72°C for 2 min.DNA polymerization proceeds along the region between them.After each cycle,the template number doubles.As each newly amplifie fragment acts as a template in subsequent cycles,successive rounds of temperature cycling can lead to an exponential increase in copy number of target DNA region.A single copy of target DNA is amplifie to up to 106copies in only 30-40 cycles in 2-3 h.PCR amplifie products are separated on an agarose gel,stained with ethidium bromide (EtBr),photographed,and the intensities of DNA bands are semi-quantified Taq DNA polymerase enables PCR a versatile tool,reducing labor,assay cost,and the potential for error while improving specificit,sensitivity and product yield and lending the assay to automation[8].Conventional PCR is a relatively easier and less expensive tool to amplify a targeted DNA fragment and useful in detection and monitoring of foodborne microorganisms.Its major disadvantages are relatively labor-intensive and time-consuming with lower detection sensitivity and accuracy,as compared to qPCR mentioned below.It is unable to distinguish viable from dead cells,and gives rise to false-positive results.
2.2.Mono-,duplex-and multiplex-qPCR methodologies
The qPCR,a modifie version of conventional PCR,has become useful for detection and quantificatio of microorganisms,owning to its high sensitivities and accuracy.Similar to conventional PCR,qPCR involves continuing amplificatio cycles during which DNA template is denatured,annealed with specifiprimers,leading to exponential increase of amplicons which are monitored in real time at every cycle with a fluores cent reporter (dye).The increased fluorescen signal is plotted against cycle threshold (Ct,i.e.cycle number) to yield amplificatio curve,from which Ct value is determined.Ct value corresponds to the cycle number for which fluorescenc signal(i.e.DNA template),which is significantl higher than background signal,is proportionally correlated with initial level of target DNA and serves as a basis for relative to absolute quantificatio of DNA template.TIn addition to all components required for conventional PCR,qPCR assays employ detection chemistries,including SYBR Green,50 exonuclease technologies,TaqMan or FRET hybridization probes for fluorescen detection ofL.monocytogenes.The increased flu orescent signal following each successive cycle enables direct quantitation of target DNA,which is correlated with template DNA concentration of bacteria present in the sample without needing post-PCR steps,making automation and high throughput possible [9,10].DNA binding dye and 5′nuclease assay are two most popular detection systems.The former is well adapted to routine analyses at low-cost while the later can allow detection of several target genes in multiplex PCR in a single reaction [10].A disadvantage of dsDNA dyes,e.g.SYBR Green,is that it can bind to all dsDNA,including PCR-amplifie products and primer dimer and thus,can potentially prevent or interfere with accurate monitoring of target sequence to be amplified Alternatively,sequence-specifiDNA probes,i.e.oligonucleotides labelled with a fluorescen dye as reporter,can enable detection only after probe hybridization with its complementary sequence,making qPCR more specific Another feature of qPCR is that for quantitative analysis,an internal amplificatio control (IAC) is included in the assay,based on the rationale that ifineither target gene nor IAC is amplified the result is invalid and the sample must be repeated.Thus,International Standard Organization(ISO)’s standard for PCR methods for the detection of foodborne pathogens(ISO22174:2005)states that“the presence of PCR inhibition shall be demonstrated using appropriate controls and that an internal amplificatio control(IAC)or external amplificatio control (EAC) should be performed with every PCR reaction”.The IAC-included qPCR assay fully meets these requirements.
Typical qPCR amplificatio is performed in a fina volume of 20-25 μL including template DNA;internal control DNA in 10×hybridization buffer with 5 mM MgCl2;PCR primers and probes specififorL.monocytogenesare added to reaction mixture and nuclease free dH2O to fina volume.The cycling parameters are usually set as follows:an incubation at 95°C for 10 min for DNA denaturation and enzyme activation,followed by 45-50 cycles consisting of a DNA denaturation step at 95°C for 10s,an annealing step (at different temperatures based on Tm of primers) for 20-30 s and an extension step at 72°C for 10 s.
The qPCR confers several advantages over conventional PCR.First,its cross-contamination risk is lower because the target presence in the sample is shown by an increased flu orescent signal;second,no post-PCR processing of amplifie DNA products is needed [11];third,fluorescen signal is proportionally linear to the level of target DNA in sample;and fourth,the assay has an automation potential [12].The qPCR is faster than conventional culture-based methods and highly sensitive,specifiand enables simultaneous detection of different target genes and/or different microorganisms [10].The qPCR has been used in identificatio and quantitation ofL.monocytogenesin clinical and food samples in a number of studies and become a more popular method forListeriadetection because high sensitivity reaching 1-10 CFU/assay in specififood matrices can be achieved although a bacterial enrichment step for at least 24 h is usually required[13-19].
The application of monoplex PCR can be limited by high cost and/or limited sample volume.To overcome these limitations and to increase PCR capacity,duplex-[17]and multiplex-[20]PCR assays,which allow simultaneous amplificatio and detection of more than one target sequences in a single reaction have been developed.In these assays,two or more primer sets targeting two or more gene sequences of the same species or different species are included in PCR mixture.With these assays,considerable time,cost and effort can be saved,and number of reactions to be performed for detecting pathogens in a food samples can be decreased [21].For development of reliable and successful duplex-and multiplex-PCR,design of appropriate primer sets is the key,as different primer sets should have similar annealing temperature (s)and the amplicons must be different in sizes with minimized interference with each other among multiple primers during amplificatio process.Additionally,appropriate and careful optimization of qPCR conditions,including the concentrations of each primer set,buffer,the balance in concentrations between dNTPs and cofactors,and DNA templates,cycling temperatures,amounts of Taq DNA polymerase and probes are also needed[22].
3.Development and applications of PCR-based assays in detection and subtyping of L.monocytogenes
Conventional PCR assays for detection ofL.monocytogenesin environmental and food samples generally include:enrichment ofL.monocytogenespresent in the samples by culturing samples in a broth (e.g.Buffered Listeria Enrichment Broth(BLEB)or Brain Heart Infusion(BHI))with selective agents and subculture toListeriaplating media;DNA extraction;speciesspecifidetection ofL.monocytogenesby target gene-based PCR;separation of the PCR-amplifie products on a agarose gel;and staining of the separated gel with EtBr,photograph and semi-quantitative analysis of amplifie DNA bands[23].
3.1.Methods for extraction and purification of bacterial DNA
Crude bacterial DNA template can be prepared as follows.About 1 mL of overnight-cultured cells ofivariousL.monocytogenesstrains in BHI broth or BLEB or enrichment samples is centrifuged at 12,000gfor 3 min.Supernatant is removed and cell pellets are washed,re-suspended in sterile distilled water or TE buffer and heated in a boiling water bath or a heat block for 10 min.The heat-treated DNA samples are cooled in ice for more than 5 min and centrifuged at 12,000gfor 10 min.The supernatant is saved as crude bacterial DNA template.About 1-5 μL of supernatant can be directly used for PCR amplifica tion[24,25].
Bacterial DNA samples can be purifie from pure culture,artificiall inoculated food samples,real food or environmental samples with DNA isolation kits.Currently,two heat-based methods,i.e.PrepMan Ultra from Applied Biosystems (Norwalk,Ct.) and InstaGene Matrix from BioRad (Hercules,Calif.),and two column-based DNA extraction methods,i.e.DNeasy Tissue Kit from Qiagen (Hilden,Germany) and UltraClean Microbial DNA isolation kit from MoBio (Carlsbad,Calif.),forL.monocytogenesare available.Other DNA isolation kits including Generation Capture Column Kit from Gentra Systems (Minneapolis,MN,USA) and Wizard DNA Clean-up System from Promega (Madison,WI,USA) are also available.Elizaquivel and Aznar [26]compared two heat-based methods and two column-based DNA extraction methods for DNA extraction from artificiall inoculated food samples.After having compared the sensitivity assays using specifiPCRs with DNA samples extracted by these methods from onion,broccoli,and green pepper artificiall inoculated with this pathogen respectively,they found that the sensitivity of DNeasy Tissue kit was the highest one among three matrices assayed forL.monocytogenes.Application of DNeasy Tissue Kit saved time and reduced overall cost of the analysis although this kit is more expensive [26].This method was used in DNA extraction fromL.monocytogenes[27-30].
3.2.Development and applications ofivarious PCR-based assays for detection of L.monocytogenes
Both conventional and qPCR assays have been applied in detection ofL.monocytogenes[23,24,31-48].The specificit and accuracy of PCR detection ofL.monocytogenesdependlargelyonselectionofappropriatetargetDNAsequences ofL.monocytogenes.To date,the target genes chosen for PCR detection ofL.monocytogenesincludedhlyAgene[13,18,23,27,34,49-52],which encodes listeriolysin O (LLO)involved in the lysis of host vacuolar membrane and is present in allL.monocytogenesstrains and essential for its full virulence [53,54],prfAgene [35-37],which encodes the central regulator for transcriptional expression of six virulence genes[55,56],iapgene [34,38,48],which encodes p60 with bacteriolytic activity essential for cell viability[57],haemolysin gene[39],which encodes a pore-forming hemolysin essential for pathogenicity [57-59],inLAB[24],which encodes internalins A and B related to entry ofL.monocytogenesinto host cells[60,61],Lm00733gene[44],which encodes an aminopeptidase[43],ssrAgene[62-64],which codes for tmRNA and functions both as tRNA and mRNA in all bacterial phyla[65]andImaA[49],which encodes for an LLO,andL.monocytogenesantigen A[66].
3.2.1.HlyA gene-based PCR assays
Thomas et al.[23]establishedhlyAgene-based PCR assay(Table1) for detection ofL.monocytogenes.PCR amplifie products with predicted size were obtained with DNA samples from 72L.monocytogenesstrains with fie different primer pairs while DNA samples fromL.ivanovii,L.innocua,L.seeligeri,L.welshimeri,L.grayi,and L.murrayistrains and 47 bacterial strains representing 17 genera were not amplified With this assay,1 pg ofL.monocytogenesDNA or 0.1 CFU of spikedL.monocytogenes/mL or g of milk and ground beef could be detected.AnotherhlyA-based 5′-nuclease qPCR assay was developed by Nogva et al.[13](Table1).In which,magnetic bead-based strategies were adapted in sample preparation and subsequent DNA purificatio with the same beads,followed byhlyAgene-based qPCR assay.This assay was positive for 65L.monocytogenesisolates but negative for 16 isolates from fie otherListeriaspecies and 18 species of other bacteria tested and could be completed in 3 h with LODs of 6-60 CFU in the linear range over 7 log units.
A key factor limitingL.monocytogenesdetection in environmental and food samples by qPCR is that qPCR assays are inhibited by food and environmental matrices,which interfere with PCR detection of foodborne pathogens.Thus,a method for physical separation of target bacteria from the competing bacteria and food matrices is needed.A method for isolation and enrichment of bacterial pathogens from food matrices employing monolayer of macrophage cells was described [67,68].Recently,Day and Basavanna [18]developed a “macrophage cell-hlyAgene-based PCR detection system” for isolation,enrichment and detection ofL.monocytogenescontaminated in infant formula using a combination of mouse macrophage-based enrichment protocol andhlyAgene-based qPCR.In this method,the mouse macrophage cells were infected withL.monocytogenespresent in infant formula.L.monocytogenesinternalized and replicated within the mouse macrophage cells.The intracellularly replicatedL.monocytogeneswere then isolated,enriched and identifie withhlyAgene-based PCR assays.With this assay,approximately 10 CFU ofL.monocytogenes/mL or g ofinfant formula could be detected after 16 h post-infection.In this assay,IAC control was included to eliminate of the possible falsenegative results.This assay was specififorL.monocytogenesas co-inoculation withL.innocuacaused no reduced sensitivity forL.monocytogenes.UsingListeriaselective media,intracellularL.monocytogeneswere isolated from infected macrophage lysates for further confirmation The macrophage enrichment method coupled with qPCR identificatio displayed a widerange ofdetection from 101to108CFU/mLofL.monocytogenesin infant formula in 20 h.Furthermore,this method was highly specifiand sensitive forL.monocytogenesin infant formula.Because of the higher susceptibility ofinfant populations to listeriosis due to their inherent immunodeficien y,rapid detection ofL.monocytogenesin infant formula is essentially needed.WhileL.monocytogenesmay initially be present in quite low quantities in foods,its multiplication during food storage can ultimately cause the increased numbers adequate to initiate disease.Indeed,L.monocytogenesin infant formula grew at cold temperatures [69].Development of this macrophage cell-hlyAgene-based PCR system for rapid detection ofL.monocytogenespresent infant formula may reduce the possibility of further consumption ofinfant formula to infant populations in the event of an outbreak and administration of appropriate antibiotics to infant victims could be initiated timely.Ahlygene-based PCR assay was developed to detect and quantifyL.monocytogenespresent in cerebrospinal flui (CSF) by Monnier et al.[70].When being used to examine a panel of bacterial species,this assay was specifiin detectingL.monocytogenes.When being applied to examine 214 CSF samples from patients suspected of having neurolisteriosis,this assay detected all the cases from whichL.monocytogeneswas isolated by culture.Furthermore,when being used to examine fie clinically confirme cases ofineurolisteriosis which were not detected by bacterial cultures,this assay was positive to all fie samples.
HlyAgene has been one of the most popularly selected targets for PCR detection and the primer/probe sets corresponding to regions of this gene were highly specififorL.monocytogenes[71].However,a disadvantage of thishlyAgene-based PCR assay is that some strains of serovar 4c were negative to this gene[49].
3.2.2.Iap gene-based PCR assay
TheiapgeneofListeriaspeciesencodesproteinp60.Bycomparingiap-related genes from differentListeriaspecies,Burbert et al.[38]identifie the common and variable regions within these genes specififor each ofListeriaspecies.Based on theseiapgene sequences,they designed PCR primers forListeriaspecies.Application of two primers from 5′-and 3′-terminal regions ofiapgenes yielded PCR products for allListeriaspecies but not for non-Listeriabacterial species.The middle section ofiapgene was highly variable betweenL.monocytogenesand otherListeriaspecies but was constant for a givenListeriaspecies.Thus,by fixin 3′-PCR primer targeting the common 3′-terminal section of theiapgenes and varying the 5′-PCR primer based on species-specifisequences from the middle section of correspondingiapgene,they were capable of distinguishingL.monocytogenesandL.innocuafromL.seeligeri,L.ivanovii,andL.welshimeri.L.monocytogenes-specifiPCR primers Mono A and Mono B(Table1)yielded PCR products representing the repeat region ofiapgene.Furthermore,the sizes of these PCR products allowed an estimation of the number of TN repeat units within the repeat region of p60 gene of aL.monocytogenesstrain as the number of repeat units differed amongL.monocytogenesisolates,enabling serotype identifica tion ofL.monocytogenesisolates.Hein et al.[14]developed and applied aniapgene-based qPCR assay to detect and enumerateL.monocytogenesandL.innocuain artificiall contaminated milk samples.A 175-bp fragment and a 309-bp fragment were amplifie fromiapgene inL.monocytogenesandL.innocua,respectively.A total of 42L.monocytogenesstrains and 33L.innocuastrains of different serovars and 22 bacterial strains not belonging to the genusListeriawere tested.When being applied in detection and quantificatio ofL.monocytogenesin milk,this assay was specififorL.monocytogenesandL.innocuawith the same sensitivity as that of traditional plate count method.Copy numbers detected with thisiapgene-based qPCR were 10-100 times lower than CFUs obtained by culture method.
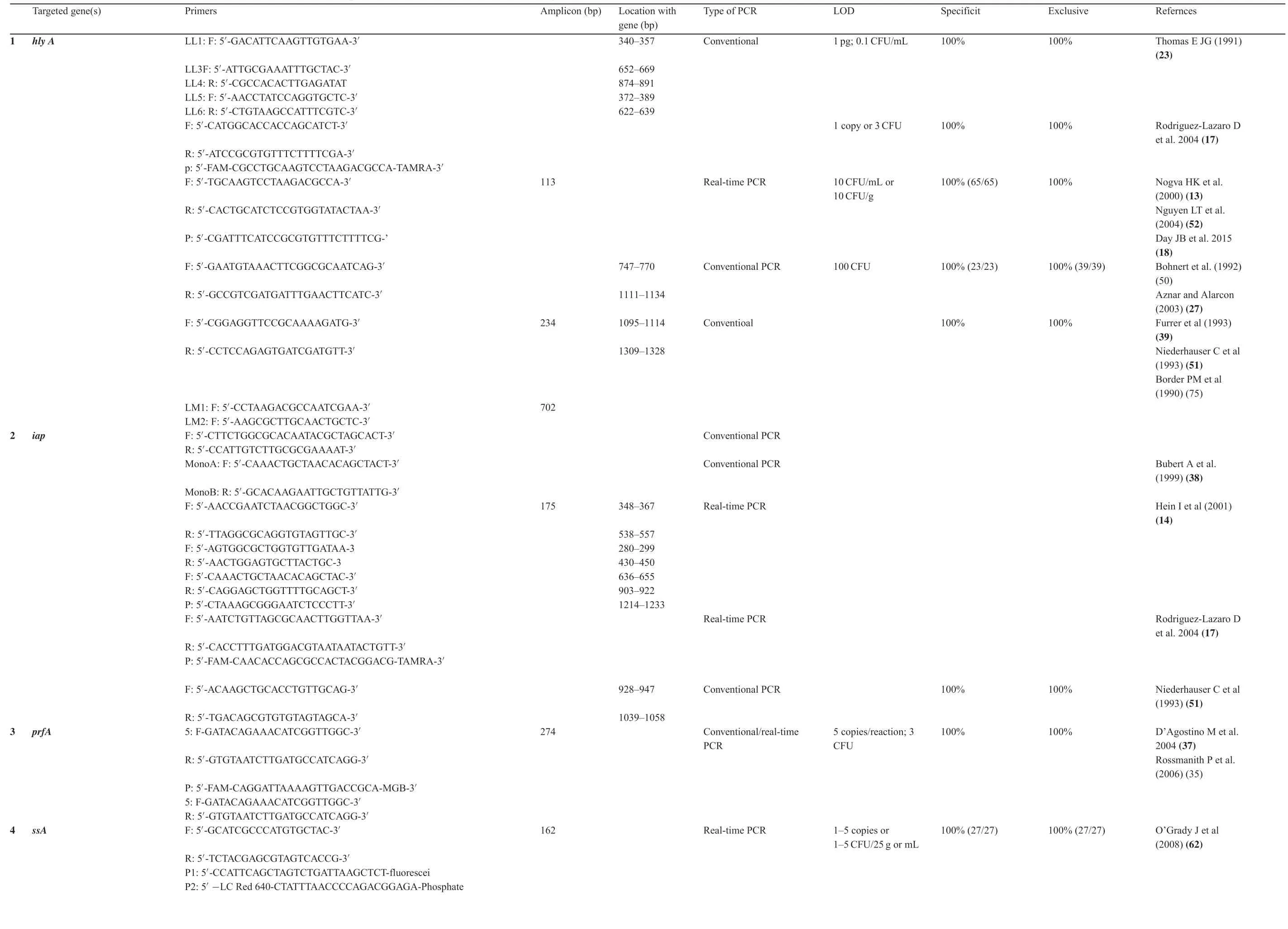
Table1 Primers and Probes used for conventional and real-time quantitative PCR for L.monocytogenes.

Table1(Continued)

?
Rodríguez-Lázaro et al.[17]compared the usefulness betweenhlyandiapgenes as targets with qPCR in term of sensitivity,specificit and accuracy for detection ofL.monocytogenes.They found that withhly-based assay,the sensitive,specifiand precise quantificatio ofL.monocytogenescould be achieved.However,withiap-based assay,heterogeneous results were generated and reliable quantificatio could be achieved only with homologous strains or strains within the same serovarrelated phylogenetic branch.
3.2.3.PrfA-based PCR assay
PrfA,a key transcriptional regulator,induces expression ofivirulence factors related toL.monocytogenespathogenesis in infected host cells.WhenL.monocytogenesenters into host cells cytosol,PrfAis post-translationally activated[72,73].PrfAhas been selected as for PCR assay targetingL.monocytogenes.For instance,D’agostino et al.[37]developed aprfA-based PCR assay coupled with an IAC,which was tested against 38L.monocytogenesstrains and 52 non-target strains.This assay was 100% inclusive and 100% exclusive.In a collaborative trial involving 12 European laboratories,this assay was validated against 14 moreL.monocytogenesstrains and 14 non-target strains with inclusivity and exclusivity being 100%and 99.4%,respectively,and with both repeatability and reproducibility being 99.4%.This assay was incorporated into a method for detection ofL.monocytogenesin raw milk after a 24 h-enrichment,followed by 16h-enrichment in a medium,which was added directly into the PCR.Its performance was assessed in a collaborative trial involving 13 European laboratories.In that trial,the specificit and sensitivity were 81.8%and 89.4%,respectively.Rossmanith et al.[35]further developed thisPrfA-based assay into qPCR assay with inclusion of IAC.Both inclusivity and exclusivity were 100% when being tested against 100L.monocytogenesisolates,30 otherListeriaspp.isolates and 29 non-Listeriaisolates with LOD being 5 copies/assay.Using a combination of enrichment and qPCR,7.5 CFU/25 mL of artificiall contaminated raw milk,and 9,1,and 1 CFU/15 g of artificiall contaminated salmon,pate,and green-veined cheese could be detected.When 76 naturally contaminated food samples were tested and compared with those obtained by ISO 11290-1 standard method,its relative accuracy,specificit,and sensitivity were 96,100,and 76.9%,respectively.
3.2.4.SsrA-,ribosomal DNA(rDNA)-,and mRNA-based PCR assays
O’Grady et al.[63]developed anssrAgene-based qPCR assay (Table1) and examined its specificit forL.monocytogenesagainst a panel of 6Listeriaspecies and 26 non-Listeriabacterial species.Its LOD was 1-10 genomic copies.When being applied in detecting naturally and artificiall contaminated culture enriched soft cheese,meat,milk,vegetables and fish its LOD was 1-5 CFU/25 mL or g of food sample in 30 h.When its performance was compared with Roche Diagnostics‘LightCycler FoodproofListeria monocytogenesDetection Kit,while both methods were able to detectL.monocytogenesin all artificiall contaminated samples,they did not detectL.monocytogenesin 27 natural retail food samples.This assay was applied for testing clinically relevant and common foodborne bacterial pathogens.Jin et al.[64]integratedssrAgene-based qPCR with high-resolution melting curve analysis in assessment of 53Listeriaspecies and 45 non-Listeriaspecies with one primer set (Table1).This assay displayed 100% accuracy relative to conventional methods and a 93.3% correction rate to 30 artificiall contaminated samples and enabled an accurate identificatio of sixListeriaspecies.Thus,it may be capable of discriminating new species for epidemiological and food safety research.
Bansal et al.[74]developed and validated a multiplex PCR assay.When it was tested in parallel with standard cultural procedures against 350 food samples,no false-positive or false-negative results were obtained.Compared to standard cultural methods,the PCR assay is highly sensitive,cost-effective and rapid with results obtained in 48 h.Border et al.[75]designed PCR assay with fie primers specificall targeting sequences fromListeriaDNA.When all fie primers were used in combination,three PCR products,including aListeriaspecifiproduct with DNA from anyListeria spp.,aL.monocytogenes-specifiproduct amplifie only in the presence of DNA from this organism and a universal product by using DNA from any bacterial source,were generated.The generation of these PCR products was used as a diagnostic to detectListeriaspp.andL.monocytogenesin foods.Lawrence and Gilmour [76]investigated the incidence ofL.monocytogenes and Listeriaspp.in poultry processing plants,raw and cooked poultry products for over a 6-month period.Within the raw and cooked poultry processing environments,36/79(46%) and 51/173 (29%) of samples containedListeriaspp.while 21/79 (26%) and 27/173 (15%) containedL.monocytogenes,respectively.Among the raw and cooked products tested,34/58(59%)containedL.monocytogenes.This multiplex PCR was a convenient and time-saving assay for rapid confirma tion ofListeriaspp.andL.monocytogenesin a single reaction[76].
Another multiplex PCR with two primer pairs was developed [77].With this assay,a 938-bp region of 16S rRNA gene highly conserved in allListeriaspecies was amplifie with the firs pair primer while a 174-bp region ofhlyAgene ofL.monocytogeneswas amplifie by the second primer pair.The isolates ofListeria spp.generated a single 938-bp product whereasL.monocytogenesisolates yielded both 938-bp and 174-bp products.When being applied to verifyListeriacolonies and to determine the incidence ofL.monocytogenes,this assay detected 57/150 (38%)mechanically separated turkey samples contaminated withL.monocytogeneswhile 18% of them harbored otherListeriaspp.Among 57L.monocytogenesisolates,51,44,and 2%were identifie to be serogroups 1,4,and others,respectively[78].
Rodriguez-Lazarro et al.[17]reported a duplex qPCR assay targeting 23S rDNA ofListeria sppandhlygenes.This method enabled efficient accurate,selective and simultaneous identificatio of 52L.monocytogenesand 120Listeriaspp.strains via FAM-labelledhlyand VIC-labelled 23S rDNA probes with specificit of one molecule being 100% for 23S rDNA and 56% forhlygene.Simultaneous and linear quantifica tion was achieved in a 5-log range,with an upper limit of 30 molecules.Somer and Kashi [79]developed another 16S rDNA-based PCR-based method.These two pair primers plus a pair primer universal to any bacterial DNA at 16S rRNA gene as a positive control in same reaction mixture were used to simultaneously detectListeria and L.monocytogenesin food products.This method can detect 1-5 CFU in a 25 g sample in≤24 h.
Additionally,multiplex reverse transcription-qPCR assays were applied to measure mRNA levels ofinlA,inlB,hly,bshandprfAgenes in 17L.monocytogenesisolates[80]and mRNA levels ofmdrL,ladR,lde,sigBandbcrABCgenes induced by benzalkonium chloride at sub-lethal concentration[81].
3.2.5.PCR assay based on novel species-specific genes for detection of L.monocytogenes
Recently,Tao et al.[82]comparatively analyzed the genomic sequences ofL.monocytogenesstrains and non-L.Monocytogenesstains and identifie 26 specifitarget sequences.They designed PCR primers targeting them for detectingL.monocytogenesstrains by comparatively analyzing the genomic sequences of strains ofL.monocytogenes andnon-L.monocytogenes.They found that among 26 genes tested,three,namelyLMOf2365_0970,LMOf23652721andmpl(Table1),were specifitoL.monocytogenes.The species-specifiprimers Lm8 for geneLMOf23650970,Lm13 for geneLMOf2365_2721and Lm20 formplgene displayed higher specificit and sensitivity with LODs being 430,43,and 4.3 fg/μL for genomic DNA,and 5×103,50,and 5 CFU/mL for pure culture ofL.monocytogeneswithout interference with specificit of detectingL.monocytogenesco-cultured with other pathogens.After 6-8 h enrichment,L.monocytogenesin milk samples artificiall inoculated with 38 CFU/10 mL could be detected with these primers.
3.3.Differentiation ofiviable from dead L.monocytogenes by qPCR in conjugation with propidium monoazide pretreatment
While qPCR has been increasingly applied in detection and quantificatio of foodborne pathogens present in food and environmental samples,two major hurdles limit their wider applications in quantitative detection of foodborne microorganisms includingL.monocytogenes.One is the detection of DNA from dead cells and difficult in distinction ofiviable from dead bacterial cells,posing a major challenge in microbial identification and diagnostics [13].Presence of DNA in environment after cells have lost their viability can be persistent.Thus,DNAbased quantificatio methods may lead to overestimation ofiviable cell numbers in mixed populations ofiviable and dead cells or even lead to false-positive results in absence ofiviable cells.Only viable cells are pathogenic.Although mRNA-based methods may circumvent this problem,they are more technically demanding and have drawbacks [83].This is a problem particular for the processed foods or long-term stored foods.This problem may be resolved by qPCR in conjugation with ethidium monoazide (EMA) or propidium monoazide (PMA)pretreatment.
EMA,being able to selectively enter damaged cells within which it can be photo-chemically cross-linked to nuclear DNA under visible light[84],was used as a way to reduce qPCR signal derived from free DNA and/or dead bacterial cells by selectively entering damaged cells and blocking DNA from being PCR amplifie via photo-activation [83,85,86].EMA-bound DNA causes a reduced qPCR signal due to inhibition of PCR amplification which can be translated to a reduced qPCR-derived dead bacterial cell counts.EMA-qPCR was applied in detection and quantificatio of different foodborne pathogens and its influenc on qPCR signal was examined [28,83,85,87].Rudi et al.[28,87]applied an EMA-qPCR approach[13]in quantitative differentiation between viable and deadL.monocytogenescells.They found that EMA-qPCR could detect <100 CFU/L ofiviable,viable but not culturable on gouda-like cheeses and that this method allowed quantificatio of >0.5% viable cells in a background of dead cells when viable cell number was above its LOD.However,the wider applications of EMA-qPCR are hinderedbythefactthatEMAitselfcanalsopenetrateintoafraction ofiviable cells of some bacterial species.While EMA is actively pumped out of metabolically active cells via membrane-bound transporters,its residue level remaining within cells can lead to substantial loss of DNA.Indeed,Flekna et al.[88]reported that EMA influence both dead and viableL.monocytogenescells and that EMA/qPCR was a poor indicator of cell viability.
This problem was resolved by Nocker et al.[89],who demonstrated that PMA was more selective than EMA in that it penetrated only into dead bacterial cells with compromised membrane but not into viable cells with intact cell membranes.Once it enters cells through compromised membrane,PMA intercalates with nuclear DNA of dead cells.Being exposed to bright light,its photo-inducible azide group allows PMA to be covalently cross-linked to nuclear DNA.This renders the DNA insoluble and causes its loss during subsequent extraction of genomic DNA.Subjecting a bacterial population comprised ofiviable and dead cells to PMA treatment can lead to selective removal of DNA from dead cells.PMA has been applied to a variety of species across the bacterial kingdom,exhibiting a major advantage over EMA.Higher positive charge of PMA than EMA might be the major reason explaining higher impermeability through intact cell membranes,thus avoiding DNA loss ofiviable cells[29,89,90].
Original protocols for PMA-qPCR [91,92]were as follows:(a) PMA cross-linking:typically,1.25 μL of 20 mol/L PMA dissolved in 20% dimethyl sulfoxide was added to 500 mL of culture aliquots to fina concentrations of 50 μM in light-transparent 1.5-mL micro-centrifuge tubes.After being incubated in the dark for 5 min with occasional mixing,sample tubes were placed about 20 cm from light source and laid horizontally on ice to avoid excessive heating and exposed to light for 2 min with a 650-W halogen light source with occasional shaking to ensure homogeneous exposure to light;(b)cells were pelleted by centrifuge and DNA was isolated and(c)qPCR assay was performed using corresponding primers and probe.Pan et al.[93]used both EMA and PMA in enumeration ofiviableL.monocytogenescells in presence of dead cells.Viable cell counts were linearly related to qPCR Ct values for PMA-treated cells from both planktonic and biofil sources within a 4-log range.Martinon et al.[30]applied PMA-qPCR to detectL.monocytogenesrecovered from artificiall contaminated food processing surfaces.Currently,PMA-hlygene-based PCR Bacterial Viability Kit forL.monocytogenesis available from Biotium.The protocols were modifie to adapt high throughput assays with 96-well plates in assays ofiviableE.coli0157H7 andSalmonella[94,95].
3.4.Serotpye-and virulence-specific determination of L.monocytogenes with multiplex PCR assays
A large proportion of human listeriosis cases are caused by infection ofL.monocytogenesserotypes 1/2a and 4b [96].Multiplex PCR Assays have been applied in serotpye-and virulence-specifidetermination ofL.monocytogenesisolates.For instance,Liu et al.[97]developed a multiplex PCR assay with primers targetinginlA,inlCandinlJgenes.Both 517 bp and 238 bp fragments could be amplifie withinlCandinlJprimer sets,respectively.The identity of 36L.monocytogenesisolates was confirme via amplificatio of an 800 bp fragment withinlAprimers.The virulence of these isolates was determined by amplificatio of 517 bp and/or 238 bp fragments.L.monocytogenespathogenic strains able to cause mortality inA/Jmice via intraperitoneal route could be detected withinlCand/orinlJprimers.However,naturally non-pathogenic strains were negative with these primers.While 8 of 10L.ivanoviistrains were detected byinlCprimers,they could be excluded as non-L.monocytogenesthrough their negative reactions withinlAprimers in multiplex PCR.Thus,application of multiplex PCR targetinginlA,inlCandinlJgenes allows simultaneous verificatio of the identity ofL.monocytogenesspecies.Burbert et al.[98]developed another multiplex PCR assay with a combination of fie different primers in a single reaction,i.e.one primer was designed based on the conserved 3′end specififor allListeriaspecies and other four primers were specififor L.monocytogenes,L.innocua,andL.grayi,or three grouped speciesL.ivanovii,L.seeligeri,andL.welshimeri,respectively.The PCR method enabled simultaneous detection ofL.monocytogenes and L.innocua[81].Borucki et al.[99]and Call et al.[100]discriminated multiple serotype and lineagespecifidifferences amongL.monocytogenesstrains by using a mixed genomic DNA microarray.Borucki and Call [101]developed a PCR-based method for identificatio ofL.monocytogenesserotype by designing Division-specifiPCR primers from variable regions ofL.monocytogenesgenome and using them in conjunction with Division III primer to classify 122L.monocytogenesstrains into fie serotype groups.The results were consistent with those of conventional slide agglutination method for 97,100,94,and 91% of strains belonging to serotypes 1/2a,1/2b,1/2c,and 4b [101].Zhang and Knabel[102]developed a multiplex PCR assay with primer sets targeting ORF2372(similar to teichoic acid protein precursor C),InlB (internalin B),inlC (internalin C) and Imo171 (putative peptidoglycan-bound protein).This assay enabled identifica tion and differentiation of serotypes 1/2a and 4b from other serotypes ofL.monocytogenes.Chen and Knabel[103]developed another multiplex PCR assay by combining detection ofL.monocytogenesserotypes 1/2a and 4b with ECs I,II,and III with primer sets targeting ECI,EciI and ECIII (Table2).This single multiplex PCR allowed determination whether or not an isolate can be genusListeriaorL.monocytogenesserotype 1/2a or 4b or ECs I,II,III.A multiplex PCR assay was established with primer sets targeting lmo1118,lmo0737,orf2110,orf2819,prs(Listeriagenus specific)pfrA(L.monocytogenesspecific andflaA[104-106](Table2).When being applied to test 187L monocytogenesstrains in Japan,99.5%of them were identifie as serotype 4b [107].Rawood et al.[108]developed a multiplex PCR with primer sets targetingprs(Imo4A 0215,phosphoribosyl pyrophosphate synthetase),isp(Im01441,putative peptidoglycan acetylation protein),L1(LMOF2365RS13380,cell wall surface anchor family protein),L2(Imo0525,hypothetical protein)andL3(LM)4A RS05595,hypothetical protein and applied it to identify genusListeriaand discriminate the major lineages among 46 isolates ofL.monocytogenes.Nho et al.[109]developed and applied a multiplex PCR with primer sets specififor serotypes 1/2c(LMOSLCC2372_0308),3a(LMLG_0742)coupled with primer sets specififor serotype 1/2a(flaA)to separate serotypes 1/2a,1/2c,3a and 3c,This assay enabled differentiation of fieL.monocytogenessubgroups,including 1/2a-3a,1/2c-3c,1/2b-3b-7,4b-4d-4e and 4a-4c,and separation of 1/2a and 1/2c strains from 3a and 3c strains.
By applying two multiplex PCR assays developed by Doumith et al.[104]and Borucki and Call[101],respectively,Camargo et al.[110]assessed the serotype distribution among 134L.monocytogenesisolates from clinical,beef,and environment samples in Brazil.They found that isolates from clinical samples were mainly serotype 4b while prevalent serotype among beef cut and environment samples was predominantly 1/2c and that results of serotyping with the protocol[101]were completely consistent with those of conventional serology.
3.5.PCR detection of L.ivanovii
BesidesL.monocytogenes,L.ivanoviiis another pathogenic species found most exclusively in ruminants.Conventional 16S rRNA gene-[111]and N-acetylmuramidase gene-based [45]PCR assays have been applied in detection ofL.ivanovii.Wanget al.[111]developed a 16S rRNA gene-based PCR with primers R-1 and R-2 and probe (Table1) that only targetL.ivanovii,but not the other species ofListeriaor other bacteria tested.This method detected all strains ofL.ivanoviitested but no other species ofListeria,including nineL.monocytogenesstrains and 20 taxonomically related bacteria.It detected 4 CFU ofL.ivanoviiin pure cultures and 4-40 CFU ofL.ivanoviiin inoculated and diluted mouse feed,blood,or fasces samples.Liu et al.[45]analyzed genomic DNA from sixListeriaspecies and isolated aL.ivanoviispecificlone containing a 946 bp insert.This clone contained twoL.ivanoviigenes and an IGS region similar to homologous regions ofL.innocuaandL.monocytogenesgenomes.A low homology region was also identifie by comparing it withL.innocua and L.monocytogenesgenomes.The designed primers amplifie a 463 bp fragment from genomic DNA ofL.ivanoviistrains only,but not those from otherListeriaspecies or common bacteria.Thus,PCR employingL.ivanovii-specifiprimers can be useful for determination ofL.ivanovii.

Table2 Primers and probes ised for PCR Genotyping.

Table2(Continued)
Rodrıguez-Lazaro et al.[112]developed a qPCR assay targetingsmcLgene for quantitative detection ofL.ivanovii.ThesmcLgene encodes a sphingomyelinase C,a membranedamaging virulence factor involved in mediating bacterial escape from phagocytic vacuole [113,114].ThesmcL-IACqPCR assay including an IAC was able to detect one genome equivalent in 45% of reactions and displayed excellent quantitative accuracy with its high linearity and efficien y over a 6-log range and 100% selectivity.It enabled detection of 2 CFU/mL of raw milk,43 CFU/mL of blood or 50 CFU/mL of amniotic flui and could be an alternative for identificatio ofL.ivanovii.More recently,Mao et al.[115]developed an assay by combining a large-volume immunomagnetic separation and multiplex PCR(Table1),which could simultaneously detectL.monocytogenesandL.ivanoviiin lettuce with LODs being 1.0 CFU/mL in pure culture and 1 CFU/g in lettuce.
3.6.PCR-based differentiation of L.monocytogenes from other listeria species
The sequences of 16S rRNA,23S rRNA,and 16S-23S rRNA IGS regions were used in subtype discrimination betweenListeriaspecies isolates [116-119].For instance,Czajka et al.[116]conducted PCR-amplificatio of 16S rDNA with primers flankin a 1.5-kb fragment.They observed the sequence differences in the V2 region of 16S rDNA betweenL.monocytogenesandL.innocuaand between differentL.monocytogenesserotypes.While bothL.monocytogenesSLCC2371 andL.innocuashared the same V2 region but differed within the V9 region.Graham et al.[117,118]characterized 16S-23S rRNA IGS regions in sixListeriaspecies and found that PCR amplificatio with a “generic primer” targeting 16S-23S IGS amplifie 340 bp (small) and 550-590 bp (large)fragments with DNA from allListeriastrains tested.SevenL.monocytogenesserotype 4b strains and oneL.monocy-togenesserotype 4d strain also had an additional 360 bp fragment from one of theseL.monocytogenesserotype 4b strains identical in DNA sequence to small 340-bp IGS.The small rRNA IGSsof L.innocua,L.ivanovii,L.seeligeri,L.welshimeri,and L.grayiwere 83%-99% homologous to that ofL.monocytogeneswhile large rRNA IGS ofL.monocytogeneswas 81%-96% homologous to otherListeriaspecies.The DNA sequences of the central 274 bp oflarge rRNA IGS of strains from sevenL.monocytogenesserotypes were highly homologous and four groups were identifie withinL.monocytogenes.
Liu et al.[44]comparatively analyzed genomic sequences ofL.innocuaandL.monocytogenesand selected aL.monocytogenes-specifilmo0733 gene for specifidetection ofL.monocytogenes.By PCR with primers targeting this gene,a 453 bp fragment was specificall amplifie only from genomic DNA ofL.monocytogenesstrains but not from otherListeriaspecies or other Gram-positive and-negative species.A real-time 5′-nuclease qPCR assay was developed and adopted as BAM protocol for simultaneous confirmatio ofL.monocytogenesandListeriaspecies isolates,includingL.innocua,L.ivanovii,L.seeligeri and L.welshimeriby targeting different regions of theiapgene and the duplex qPCR assay is intended for the confirmatio of onlyL.monocytogenesisolates[120].
L.monocytogenesisolates representing eight EcoRI ribotypes were differentiated into 10sigBallelic types [121],implying thatsigBsequencing can provide discriminatory power and allow subtype discrimination within eachListeriaspecies.Characterization ofL.monocytogenesisolates by both MLST(sigB,actA,gap,inlA,purM,prs,ribC,and purM,) and automated EcoRI ribotyping [122]showed the improved subtype discrimination at a much higher cost per isolate.Sauders et al.[123]reported that PCR amplification/sequencin ofsigBgene could be applied in species-level identificatio and subtyping ofListeriaisolates.Comparison ofsigBsequences with the Food Safety LaboratorysigBallelic type database allowed classifica tion to species and characterization to allelic type.An allelic type was define as a distinct sequence for the 660-bpsigBfragment;two isolates were assigned a different allelic type if they differed by at least one nucleotide among 660 nucleotides[124].Simmon et al.[124]characterized 245 putativeListeriaspp.isolates bysigB-based PCR and classifie them into three species,includingL.innocua,L.seeligeriandL.welshimeri.These finding are in agreement with one thatL.innocuawas the predominantListeria spp.in urban environments[123].ThesigBsequence data provided an chance for initial subtype discrimination among theListeria spp.isolates.Overall,17sigBallelic types were differentiated among the 249Listeriaspp.isolates characterized,with eight,fie,and four allelic types being found amongL.innocua,L.seeligeri,andL.welshimeriisolates.This approach can be used to confir isolates asListeriaspp.;classify isolates to species level and provide rapid,economical,and reliable species identification Hage et al.[125]developed two triplex PCRs with one identifyingL.monocytogenes,L.seeligeri,and L.welshimeriand the other identifyingL.ivanovii,L.grayi and L.innocua.The primers and probes were designed targeting genes specififor L.seeligeri[126],L.welshimeri[46],L.ivanovii[45],L.innocua[127],andL.grayi[128](Table2).All theListeriastrains were identifie as expected species byamplificatio ofthe predictedtargets withCt valuesrangingfrom15to30.DNAsamplesfrom53non-ListeriaGram-positive bacteria and 49Listeriaspp.derived from human,animal,food or food-processing environments were evaluated.The assay could identify each of six species from a mixture of strains.Thus,it can be a powerful method implemented in specialist reference laboratories for rapid identificatio ofListeria species[125].
4.Application of droplet digital PCR(ddPCR)in detection of L.monocytogenes
While qPCR methodologies have been widely applied in detection ofL.monocytogenes,a major problem is that PCR amplificatio is frequently inhibited by a variety of organic and inorganic substances present in environmental,biological,and food samples,leading to a decreased sensitivity and even totally false-negative results.The inhibitory substances may also be unintentionally introduced during transport,processing or DNA extraction steps[129].This problem seriously limits wider applications of qPCR in detection ofL.monocytogenesin environmental and food sources,calling for new methodologies.The ddPCR may provide a partial solution to this problem.
The ddPCR technology is a digital PCR employing a wateroil emulsion droplet system.The “droplets” are generated in a water-oil emulsion to form the partitions capable of separating template DNA molecules.The droplets act the same role as individual test tubes/wells.ddPCR system partitions DNA samples into millions of droplets,which support PCR amplificatio of template molecules.After PCR,end-point PCR amplifie product is detected by fluorescenc intensity of probes and each droplet is analyzed or read in a f ow cytometer to determine the proportions of PCR-positive droplets in original sample to determine the level of target DNA template in original sample by Poisson statistics.ddPCR workflw includes droplet generation,thermal cycling,droplet reading,and data analysis[130].
Doi et al.[131]compared the effectiveness between ddPCR and qPCR in detecting environmental DNA(eDNA)from sunfis in ponds with two different PCR reagents.They found that ddPCR had higher detection rates of eDNA in pond water than with qPCR,especially at low DNA concentrations.ddPCR displayed a detection rate higher than that of qPCR,suggesting that ddPCR is more resistant to PCR inhibitors than is qPCR.ddPCR appears to outperform qPCR for detecting eDNA(environmental DNA),especially in habitats with PCR inhibitors.Bian et al.[132]developed a novel ddPCR assay for detection ofL.monocytogenesandE.coliO157.In this assay,a mineral oil-saturated polydimethylsiloxane (OSP) chip was used to overcome droplet evaporation.The droplet generation function,on-chip amplificatio and end-point fluorescenc readout were integrated.Two different types of bacteria were simultaneously detected with differentially labeled TaqMan-MGB fluorescen probes.Compared with qPCR approach,OSP chip-based duplex ddPCR platform exhibited higher sensitivity at the level of single molecule resolution without significan cross-assay interference.Moreover,its LOD was 10 CFU/mL for both bacteria in 2 h.The ddPCR was applied to quantifyL.monocytogenesstrains at different times during biofil formation,and to study anti-adhesive actions ofinatural bioactive substances.The results showed that the approach might be applied for adhesion assays and biofil research[133].
ddPCR is claimed to possess advantages over qPCR,including absolute quantification unparalleled precision,increased signal-to-noise ratio,removal of PCR bias,reduced consumable costs,lower equipment costs and superior partitioning.However,ddPCR does have a limitation in quantifying high numbers of target molecules[134]and a narrower range than qPCR.Its upper quantitation limit is several orders of magnitude lower than that of qPCR[135].
5.Applications of PCR-based methodologies in detection and genotyping of L.monocytogenes in food and environmental sources
5.1.Detection and genotyping of L.monocytogenes in milk and dairy products
A number of foodborne outbreaks oflisteriosis have been linked to increased consumption of contaminated dairy products[136,137].In several countries,raw milk is sold as a RTE(Ready to Eat) food after packaging [137-140].An outbreak oflisteriosis occurred in Austria and Germany caused by consumption of Quargel cheese,including 14 cases (5 deaths) by infection of a serotype 1/2a(EC 1).Serotype 1/2a(EC2)spread by this product was linked to 13 cases(two deaths)in Austria and six(one death)in Germany[137-139].About 6.4%of European red cheeses samples was contaminated withL.monocytogenes[141]andL.monocytogenesprevalencelevelswere4.9and7.0%in bulk milk collected from 474 herds in three PacifiNorthwest states in U.S.[142].Yang et al.[143]used carboxyl modifie magnetic nanoparticles to bind rabbit anti-L.monocytogenesvia amine groups,combined nanoparticle-based immune-magnetic separation (IMS) with qPCR and then applied to detectL.monocytogenesfrom artificiall contaminated milk.With this combination,L.monocytogenesDNA was detected in milk samples with LOD of ≥102CFU/mL ranging from 102to 107CFU/mL.The cell numbers ofL.monocytogenescalculated from Ct values were 1.5-7 times lower than those counted from culture plate.Hein et al.[14]developed and applied aniapgene-based qPCR assay in direct detection and enumeration ofL.monocytogenesandL.innocuain artificiall contaminate milk samples.A 175-bp (forL.monocytogenes) and a 309-bp(forL.innocua) DNA fragments were amplified Its specifici ties toL.monocytogenesandL.innocuawere tested with 42L.monocytogenesstrains and 33L.innocuastrains belonging to different serovars and confirme by using 22 bacterial strains not belonging to the genusListeria.Six copies ofiapgene per PCR could be detected using purifie DNA.When applied in detection and quantificatio ofL.monocytogenesin milk samples,copy numbers detected by this assay were 10-100 times lower than CFUs obtained by plate count method and as sensitive as traditional plate counting.Using both biochemical and PCR tests,Jamali et al.[144]investigated the prevalence ofListeriaspecies in 446 raw milk samples collected from farm bulk milk tanks and characterized them.They found thatListeriaspp was present in 22.5,16.4 and 4.9% of raw cow milk,sheep milk and goat milk samples with an overall 18.6%among all the samples examined.L.monocytogenesaccounted for 21.7%,and 18L.monocytogenesisolates were distributed into “1/2a,3a”,“1/2c,3c”,and “4b,4d,4e”.All theL.monocytogenesexamined contained internalin genes(inlA,inlC,andinlJ) and substantial proportions were resistant to tetracycline and penicillin G.Jamali et al.[25]also detectedListeriaspp.in 21 out of 207 bovine mastitis milk samples from dairy farms in Iran with 17L.monocytogenesisolates being genotyped into serogroups‘4b,4d,4e’,‘1/2a,3a’,‘1/2b,3b,7′and‘1/2c,3c’.They all harbored virulence genesinlA,inlCandinlJ.
TaqMan-based qPCR assays targetingiapgene[14],listeriolysin/hly[13,28],hlyOgene [143,145],iap/hlyOgene [146],prfA [35,139],IRS of 16S-23S rRNA genes [147]andactA[148]have been applied in detection ofL.monocytogenesin milk and dairy products.For instance,Hein et al.[14]applied aiapgene-based qPCR in direct detection and enumeration ofL.monocytogenesandL.innocuaby amplify a 175 bp (L.monocytogenes) and a 309 bp (L.innocua) fragments in the artificiall contaminated milk samples.They tested 42 strains ofL.monocytogenesand 33 strains ofL.innocuabelonging to different serovars and confirme its specificit by testing 22 bacterial strains not belonging to genusListeria.Six copies ofiapgene per PCR could be detected using purifie DNA and that these bacterial cells in a range over 5 log units could be linearly quantified Furthermore,when being applied in direct detection and quantificatio ofL.monocytogenesin milk,copy numbers detected were one to two logs higher than CFUs by plate count and as sensitive as traditional plate counting method.L.monocytogenesisolated from 6.5%(56/861) of bulk tank milk samples were distributed among 5 serotypes(1/2a,1/2b,3b,4b,and 4c)with 93%being serotypes 1/2a,1/2b,and 4b,the most common human clinical isolates[149].
5.2.Detection and genotyping of L.monocytogenes in meats,meat products and meat-processing environment
Zhang et al.[150]characterizedL.monocytogenesisolates recovered from retail RTE-meats,fresh produce and raw chickens by serogroup identificatio via PCR,genotyping and antimicrobial susceptibility testing.They identifie fieL.monocytogenesserogroups among the 167 isolates as follows:68 (41%) as serogroup 1/2b and 3b;53 (32%)as serogroup 4b,4d,and 4e;43 (26%) as serogroup 1/2a,3a;2 (1.2%) as serogroup 1/2c,3c;and 1 (0.6%) as serogroup 4a,4c.A large proportion ofL.monocytogenesisolates were resistant to sulfonamide while some isolates were resistant to tetracycline and ciprofloxacin Tet (M)gene was detected in 14 tetracycline-resistant isolates.Isolates resistant to one or more antibiotics was commonly found.
Wieczorek et al.[78,151,152]tested the presence ofL.monocytogenesin 417 retail beef meat samples collected from the eastern part of Poland during October 2009 to January 2011.They reported that 19.4% were positive forL.monocytogenesidentifie by both culture-based and PCR-based methods.Molecular serotyping by PCR revealed that 61.7%and 32.1% ofL.monocytogenesstrains were classifie as 1/2a and 1/2c serotype,respectively,and only fie strains belonged to serotypes 1/2b (4 isolates) or 4b (one isolates).All the isolates containedinlA,inlC,inlJ,andlmo2672,while two strains of 4b serotype ofL.monocytogeneshad another virulence marker gene-llsX and 17.3% ofisolates were resistant to ceftriaxone.Wu et al.[153]reported that among 248 isolates ofL.monocytogenesfrom in raw food samples in China,45.2% belonged to serogroup 1/2a-3a,30.6% to 1/2b-3b,followed by 1.2c-3c (16.1%),4b-4d-4e (5.2%) and 4a-4c(2.8%).Fang et al.[154]characterized the prevalence and virulence ofL.monocytogenesisolates in chilled pork in Zhejiang province,China.They found that 196 out of 331 meat samples tested were positive forListeriaspp.withL.monocytogenesaccounting for 11.5% and that 60.5% ofL.monocytogenesserotype belonged to 1/2c,followed by serotypes 1/2a(28.9%),1/2b(7.9%),and 4b(2.6%).All theL.monocytogenesisolates contained virulence-associated genes examined.Together,these results indicate thatL.monocytogenesdetected in pork meats and raw beef possessed virulence markers making them potentially pathogenic for humans and that resistance to one or more antibiotics among these isolates is common.Since serotype 4b has been most frequently linked to human listeriosis,its potential presence in these foods can be an important public health concern.
5.3.Detection of L.monocytogenes and L.ivanovii from seafood,seafood products and processing environment
L.monocytogenesis widely distributed in fresh water,coastal sea water and live fis in general environment.In fact,its presence has been detected in water bodies,fish shellfish lightly preserved seafood (e.g.smoked seafood products,Gravad products,seafood salad and fis roe),RTE refrigerated seafood products,seafood processing environments [155].As lowas <100 CFU/g ofL.monocytogenesare frequently found in seafood due to contamination or recontamination possibly occurring during processing [156].Approximately 1600 incidences ofL.monocytogenesderived from seafood products have been implicated in human listeriosis outbreaks[155].PCR and PCR-based genotyping methodologies have been applied in detection and characterization ofL.monocytogenesin seafood,seafood products and processing environment.
Norton et al.[157]applied PCR-based method and DNA fingerprintin methods to investigate the ecological prevalence ofL.monocytogenesamong 531 samples,including raw fis and cold-processed fish finishe product,and environmental samples collected from smoked fis processing facilities.A total of 95 (17.9%) of the samples tested were positive forL.monocytogenesusing BAX (DuPont USA) for screeningListeria monocytogenes,including 27.7%(57/206)environmental samples,7.8%(8/102) raw material samples,18.1% (23/127)samples from fis during various stages of processing and 7.3%(7/96) finishe product samples.L.monocytogeneswas isolated from 85 samples (16.0%) using culture-based methods.Used in conjunction with 48-h enrichment inListeriaEnrichment Broth,PCR system had a sensitivity and specificit of 91.8%and 96.2%,respectively.Thimothe et al.[158]characterize contamination patterns ofL.monocytogenesin RTE seafood production environments in smoked fis processing plants with 234 raw fish 233 finishe products,and 553 environmental samples.L.monocytogeneswas isolated from 3.8,1.3 and 12.8%of raw fish finishe product,and environmental samples,respectively.Among environmental samples,L.monocytogeneswas detected in 23.7,4.8,10.4 and 12.3% of samples from drains,food contact surfaces,and employee contact surfaces,and other nonfood contact surfaces,respectively.Overall,L.monocytogenesprevalence in plant environment showed a significantl positive relationship with its prevalence in finishe product samples.Rodríguez-Lázaro et al.[159]evaluated eight different pre-PCR sample processing strategies combined with a qPCR assay for detection ofL.monocytogenesin salmon products.The optimal pre-PCR procedure involved filtratio and DNA purification This strategy could detect 10 CFU/gof smoked salmon and quantifie 1000 CFU/g with excellent accuracy.Wuet al.[153]reported that 7.1%(20/280)seafood,freshwater fis and shrimp were positive toL.monocytogeneswith 4(20%),2(10%),3(15%),8(40%)and 3(15%)being identifie as subtypes 1/2a-3a,1/2c-3c,4b-4d-4e,1/2b-3b-7,and 4a-4c,respectively.Klan et al.[160]appliedhlyAgene-based PCR to analyze 150 fis meat samples collected in India and found that 4.0%of fis meats were contaminated withL.monocytogenes.Momtaz and Yadollahi[161]characterized 18L.monocytogenesisolates from 300 fresh seafood samples collected in supermarkets in Iran and detected 4b,1/2a and 1/2b serotypes among 66.7,5.6 and 27.8 (%) of these isolates,respectively and the virulence genes includingplcA,prfA,actA,hlyA and iapin all 18L.monocytogenesisolates.Jamali et al.[144]investigated prevalence,antimicrobial susceptibility and virulent typing ofL.monocytogenesamong 862 raw fis samples collected from open-air fis markets in Northern region of Iran.They observed that 32.3%and 14.3%of these samples were positive toL.monocytogenes and L.ivanovii.Approximately 72.1% (31),23.3%(10)and 4.7%(2)of 43L.monocytogenesisolates were identifie as serovars 1/2a,4b,and 1/2b.The virulence-related genes(inlA,inlB,inlC,inlJ,actA,hlyA,iap,plcA,and prfA)were detected in these isolates and substantial proportions of which were resistant to antibiotics including tetracycline,ampicillin,cephalothin,penicillin G,and streptomycin.
6.Summary and prospective
In this review,we provided a relatively complete overview of development and application ofivarious PCR-based methodologies targeting genes specififorListeria spp.,includingL.monocytogenes and L.ivanovii.They can be applied in combination with other methodologies in detection,characterization and subtyping ofL.monocytogenesin various types of food and environmental sources.However,qPCR inhibition by various inhibitors present in food matrices and environmental samples and the requirements of expensive equipment/reagents and highly technical demand for their operation are the important hurdles limiting their wider applications in detectingL.monocytogenesin food and environmental sources.Under certain circumstances,otherqPCRmethodscanbechosenifonemethod does not work for whatever reason(s).Furthermore,additional improvement of these qPCR technologies is needed to completely overcome the issue of qPCR inhibition.On the other hand,new technologies,such as bacteriophage-based assays[162-164],can also be explored as a potentially promising alternative.
Disclaimer
Some reagents and other research tools described here are available from commercial sources.We are not recommending a particular product,but only providing an example of where it might be available.The authors have no conflic ofinterest with the companies mentioned in this review.We do not stand for officia FDA or Maryland DHMH opinions of Food,biologic,drug or device products mentioned in this article.
Conflicts ofinterest
None.
Acknowledgements
Due to limited space we apologize to our colleagues that some of their invaluable works could not be included in this publication.Research described here was supported in part by FDAIntramuralChiefScientifiChallengeGrantandOakRidge Institute for Science and Education U.S.A.

- 食品科学与人类健康(英文)的其它文章
- Potential antioxidant and cytoprotective effects of essential oil extracted from Cymbopogon citratus on OxLDL and H2O2 LDL induced Human Peripheral Blood Mononuclear Cells(PBMC)
- Extraordinary tunable dynam ic range of electrochem ical aptasensor for accurate detection of ochratoxin A in food samples
- Dietary fenugreek(Trigonella foenum-graecum)seeds and garlic(Allium sativum)alleviates oxidative stress in experimental myocardial infarction
- α-Glucosidase inhibitor produced by an endophytic fungus,Xylariaceae sp.QGS 01 from Quercus gilva Blume
- About the Beijing Academy of Food Sciences
- GUIDE FOR AUTHORS