
氯冉酸荷移分光光度法测定尼扎替丁的含量
2018-03-29 05:10廖羽,余兰
遵义医科大学学报 2018年1期
廖 羽,余 兰
(遵义医学院 药学院分析化学教研室,贵州 遵义 563099)
尼扎替丁(Nizatidine)化学名为N-[2-[[2-(二甲胺基甲基)-4-噻唑基]甲硫基]乙基]-N’(-甲基-2-硝基-1,1-乙烯二胺),是一种新型强效H2受体拮抗剂,作用于胃酸分泌细胞,阻断胃酸形成并能使基础胃酸降低,亦可抑制食品和化学刺激所致的胃酸分泌[1]。
目前,测定尼扎替丁的方法主要有超高效液相色谱串联质谱法[2]、高效液相色谱法[3-5]、荧光探针法[6]、紫外分光光度法[7]等。其中,超高效液相色谱串联质谱法、高效液相色谱法检测限最低,分析成本高;荧光法灵敏度高,但不易筛选合适的荧光探针。而分光光度法,仪器价格相对低廉,操作简便,对操作人员的技术素质要求较低,方法容易进行推广。
电荷转移络合物[8](简称荷移络合物)是由电子给体和电子受体之间通过电荷转移而生成的荷移络合物。荷移络合物具有特定的颜色和吸收波长,荷移络合物的快速形成使得它在有机分子和药物分子中的定量分析得到运用。Darwish等[9]用氯冉酸(p-chloranilic acid :PCA)电子受体测定克唑替尼(化学名:((R)-3-(1-(2,6-二氯-3-氟苯基)乙氧基-5-(1-(哌啶-4-基)-1H-吡啶-4-基)吡啶-2-胺)的含量;Elqudaby.H.M等[10]用四氰基乙烯、2,3-二氯-5,6-二氰基对苯醌、7,7,8,8-四氰基苯醌二甲烷电子受体测定盐酸洛哌丁胺(化学名:N,N-二甲基-α,α-二苯基-4-(对氯苯基)-4-羟基-1-洛哌丁胺盐酸盐)和曲美布汀(化学名:2’-苯基-2’-二甲氨基正丁醇)的含量。荷移分光光度法除具有灵敏度高、分析速度快、简便等优点外,最主要的原因是对不吸收可见光的无色物质可以用受体试剂反应变成有色物质,使之能用紫外-可见分光法测定,而且能提高测定灵敏度和选择性。
易跃能等[7]用紫外分光光度法在波长314 nm处测定尼扎替丁的含量,此方法线性范围是2.506~20.05 μg/mL。荷移分光光度法测定尼扎替丁的含量鲜见报道。本文利用尼扎替丁是富电子体的化合物,氯冉酸是缺电子的醌类化合物,两者在一定条件下发生荷移反应,形成新的荷移络合物,从而建立一种测定尼扎替丁含量的新方法。与其它检测方法相比较,该方法不仅简单方便,灵敏快速,而且还有效避免了常见药物辅料和无机离子的干扰。
1 材料与方法
1.1 仪器 TU-1900双光束紫外可见分光光度计(北京普析通用仪器有限责任公司);AL204电子天平(梅特勒托利多上海有限公司);恒温水浴锅HH-ST型(郑州长城科工贸有限公司)。
1.2 材料与试剂 尼扎替丁标准品(中国食品药品检定研究院,批号:100853-200601);氯冉酸(Alfa Aesar);尼扎替丁分散片(威特药业有限公司);尼扎替丁胶囊(天津君安生物制药有限公司);乙腈、甲醇、无水乙醇、二氯甲烷、1-正丙醇、二甲亚砜、95%乙醇(分析纯,成都市科龙化工试剂厂),实验用水为去离子水。
1.3 溶液的配制
1.3.1 尼扎替丁工作溶液 精密称取10.0 mg尼扎替丁标准品,加乙腈溶解于25 mL量瓶中并稀释至刻度,摇匀。即得质量浓度为0.4 mg/mL的尼扎替丁储备液,4 ℃冰箱内保存。
1.3.2 氯冉酸溶液 精密称取50.0 mg氯冉酸,加乙腈溶解于50 mL量瓶中并稀释至刻度,摇匀。即得质量浓度为1.0 mg/mL的氯冉酸溶液,现配现用。
1.4 荷移反应条件优化
1.4.1 测定波长的选择 取适量的尼扎替丁于5 mL比色管中,加入1.0 mg/mL的氯冉酸溶液1.0 mL,用乙腈稀释至刻度,摇匀,以试剂空白为参比,用1 cm比色皿在350~600 nm范围内对尼扎替丁、氯冉酸原溶液和荷移络合物溶液进行光谱扫描,绘制吸收光谱图。
1.4.2 溶剂的选择 分别选用甲醇、无水乙醇、水、二甲亚砜、乙腈、二氯甲烷、1-正丙醇以及95%乙醇为溶剂,考察不同溶剂对体系物吸光度的影响。
1.4.3 表面活性剂的影响 选用十六烷基硫酸钠、十六烷基三甲基溴化铵、乳化剂、吐温-80、聚氧乙烯蓖麻油EL-40、氧化蓖麻油聚氧乙烯醚RH-40作为表面活性剂,考察表面活性剂对体系吸光度的影响。
1.4.4 氯冉酸用量的影响 固定尼扎替丁的质量浓度,改变氯冉酸的用量为0.2、0.5、0.8、1.0、1.5、2.0 mL,考察氯冉酸用量对体系吸光度的影响。
1.4.5 反应温度的选择 在其它条件不变的情况下,改变反应温度为20、25、30、35、40、45、50 ℃,考察反应温度对体系吸光度的影响。
1.4.6 反应时间的选择 在其它条件不变的情况下,改变反应时间为0、5、10、20、30、40、50、60、90 min,考察反应时间对体系吸光度的影响。
1.5 实验方法学考察
1.5.1 线性关系和检测限(LOD)、定量限(LOQ)考察 精密量取0.4 mg/mL的尼扎替丁对照品溶液0.2、0.4、0.6、0.8、1.0、1.5、2.0、2.5、3.0 mL,分别置于5 mL比色管中,按“1.4”项下荷移反应条件优化后的方法分别测定其吸光值。以浓度为横坐标,吸光度为纵坐标,绘制标准曲线。LOD和LOQ的计算基于响应值的标准偏差和标准曲线的斜率,即LOD=3.3δ·S-1,LOQ=10δ·S-1(δ是截距的标准偏差,S是标准曲线的斜率)得到。
1.5.2 干扰物的影响 固定体系中尼扎替丁浓度为176.50 μg/mL,考察常见药物赋形物和常见干扰无机离子对体系吸光度的影响。
1.5.3 样品含量测定 取尼扎替丁分散片10片(标示量为0.15 g/片)或尼扎替丁胶囊10粒(标示量为0.15 g/粒),精密称定,研细,精密称取适量(约相当于尼扎替丁45.0 mg),置100 mL量瓶中,加乙腈使尼扎替丁溶解并稀释至刻度,摇匀,滤过,即得2种制剂的样品溶液。取2种样品溶液各1.0 mL,按“1.4”项下荷移反应条件优化后的方法分别测定2种药物中尼扎替丁的含量,并与高效液相色谱法测得的含量结果相比较。
1.5.4 重复性试验 按“1.5.3”项下方法平行制备6份尼扎替丁样品溶液,按“1.4”项下荷移反应条件优化后的方法测定吸光度,计算RSD值。
1.5.5 日间精密度试验 精密吸取“1.5.3”项下尼扎替丁样品溶液,连续3 d,按“1.4”项下荷移反应条件优化后的方法测定吸光度,计算RSD值。
1.5.6 稳定性试验 取“1.5.3”项下尼扎替丁样品溶液,在室温下放置,分别于5、10、30、60、90、120、180、240 min后按“1.4”项下荷移反应条件优化后的方法测定吸光度,计算RSD值。
1.5.7 耐用性试验 取尼扎替丁分散片10片(标示量0.15 g/片),精密称定,研细,精密称取93.0 mg,置50 mL量瓶中,加乙腈使尼扎替丁溶解并稀释至刻度,摇匀,滤过,即得样品溶液。取1 mL样品溶液至5 mL比色管中,微小改变荷移条件,测定其含量。
1.5.8 加样回收试验 分别取“1.5.3”项下尼扎替丁样品溶液1.0 mL于5 mL比色管中,精密加入相当于尼扎替丁含量80%、100%、120%对照品溶液,按“1.4”项下荷移反应条件优化后的测定方法进行回收试验。
1.6 络合比测定
1.6.1 摩尔连续变化法 固定尼扎替丁(C1)和氯冉酸(C2)的总的摩尔浓度不变,连续改变尼扎替丁和氯冉酸之间的比率,配制一系列溶液,以试剂空白为参比,分别测定吸光度。以C1/(C1+C2)为横坐标,吸光度为纵坐标,作图,得摩尔连续变化曲线图。
1.6.2 摩尔比法 固定尼扎替丁(C1)的浓度不变,不断改变氯冉酸(C2)的用量,使尼扎替丁与氯冉酸的比例从4∶1到1∶4的改变,配制一系列溶液,以试剂空白为参比,分别测定吸光度。以氯冉酸用量为横坐标,吸光度为纵坐标,作图,得摩尔比法曲线图。
2 结果
2.1 荷移反应条件优化结果 由图1可知,尼扎替丁在350~600 nm范围内没有吸收,氯冉酸的最大吸收波长是432 nm,尼扎替丁-氯冉酸络合物的最大吸收波长是518 nm。故本实验选择518 nm为测定波长。在乙腈介质中,体系吸光度最大,且表面活性剂对体系没有增敏作用。当氯冉酸的用量为1.0 mL时,体系吸光度达到最大,随着氯冉酸用量的增加,体系的吸光度基本不变,所以本实验氯冉酸的用量采用1.0 mL。反应温度对荷移反应没有影响。尼扎替丁与氯冉酸在室温放置10 min后,体系吸光度达到最大值,且在1.5 h内吸光度基本不变。即本实验的测定方法是:以乙腈为介质,氯冉酸加量为1.0 mL,在室温放置10 min,以试剂空白为参比,于518 nm处测定络合物的吸光度。
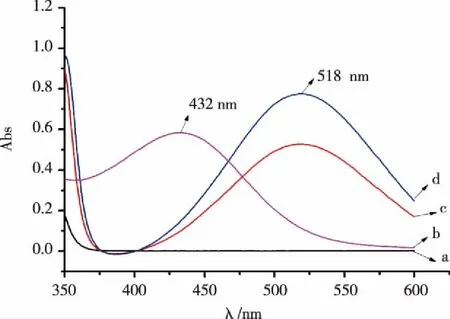
A:尼扎替丁原溶液;B:氯冉酸原溶液;C:133.00 μg/mL的尼扎替丁+氯冉酸溶液;D:200.00 μg/mL的尼扎替丁+氯冉酸溶液。图1 尼扎替丁、PCA及荷移络合物吸收光谱图
2.2 尼扎替丁的含量测定
2.2.1 线性关系和检测限(LOD)、定量限(LOQ)考察 以尼扎替丁浓度为横坐标(C),吸光度为纵坐标(A)绘制标准曲线,回归方程为A=0.003 9c+0.018 98(r=0.999 7),尼扎替丁浓度在16.00~240.00 μg/mL范围内与吸光度呈线性关系。摩尔吸光系数ε=1.80×103L/(mol·cm)-1。LOD=3.22 μg/mL,LOQ=9.77 μg/mL。
2.2.2 干扰物的影响 当相对误差在±5%范围内时,1000倍的葡萄糖、蔗糖,500倍的淀粉、乳糖、环糊精,200倍的Na+、Cl-,50倍的聚乙二醇,10倍的羟丙基甲基纤维素、K+,5倍的Ca2+,2倍的柠檬酸物质对测定无干扰。
2.2.3 样品含量测定 将“1.5.3”项下的样品溶液进行测定和计算,结果见表1。结果表明,本法的测定结果和文献方法测定结果相吻合。
表1尼扎替丁制剂中含量测定结果(n=3)

样品本文方法(%)HPLC法(%)[5]分散片99.55±0.6299.51±0.95胶囊99.62±0.55100.67±0.20
2.2.4 重复性、日间精密度、稳定性试验 根据各测定结果计算,重复性、日间精密度、稳定性的RSD值分别为0.62%、0.82%、0.82%,表明本方法精密度良好,络合物溶液在室温下4 h内基本稳定。
2.2.5 耐用性 将“1.5.7”项下的样品溶液进行测定和计算,结果见表2。结果表明,本法耐用性良好,符合测定要求。
表2耐用性测定结果(n=3)
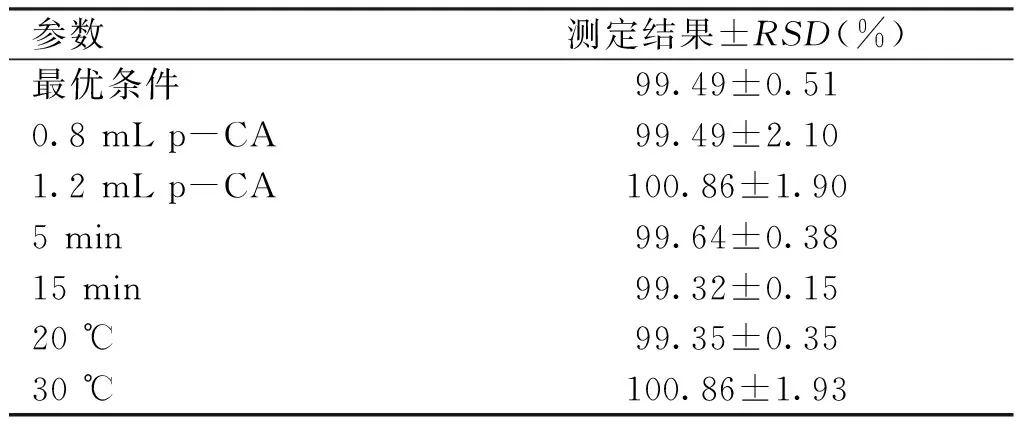
参数测定结果±RSD(%)最优条件99.49±0.510.8mLp-CA99.49±2.101.2mLp-CA100.86±1.905min99.64±0.3815min99.32±0.1520℃99.35±0.3530℃100.86±1.93
2.2.6 加样回收试验 将“1.5.8”项下的样品溶液进行测定和计算,结果见表3。结果表明,本法的准确度较高,数据可靠,可用于药物制剂中尼扎替丁含量的测定。
表3回收率试验结果(n=3)
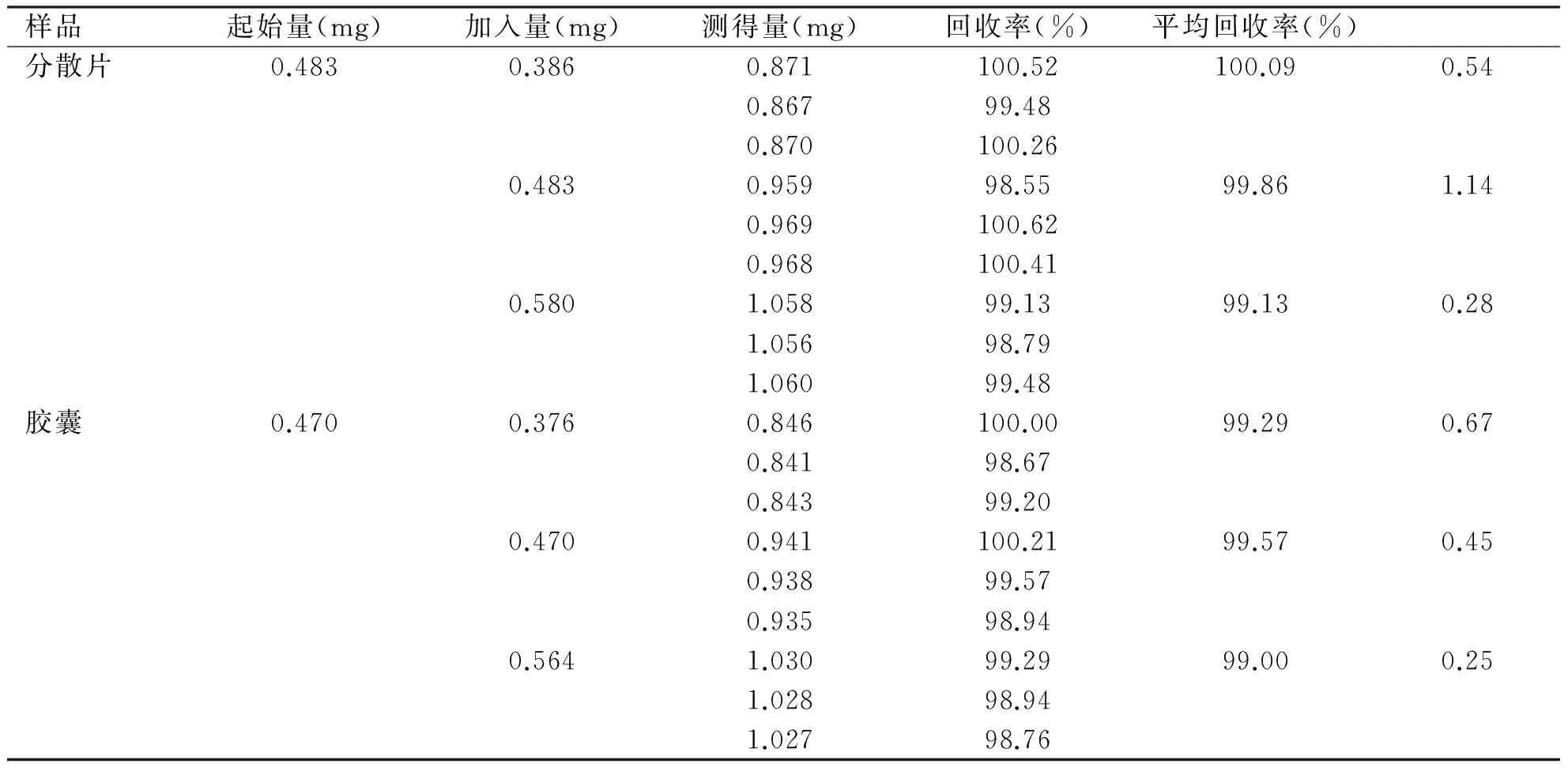
样品起始量(mg)加入量(mg)测得量(mg)回收率(%)平均回收率(%)分散片0.4830.3860.871100.52100.090.540.86799.480.870100.260.4830.95998.5599.861.140.969100.620.968100.410.5801.05899.1399.130.281.05698.791.06099.48胶囊0.4700.3760.846100.0099.290.670.84198.670.84399.200.4700.941100.2199.570.450.93899.570.93598.940.5641.03099.2999.000.251.02898.941.02798.76
2.3 络合比测定结果 运用等摩尔连续变化法(见图2)和摩尔比法(见图3)测得氯冉酸与尼扎替丁之间的荷移络合物的组成比为1∶1,络合常数为1.8×104L/mol。
氯冉酸是一平面缺电子结构,可作为电子受体,尼扎替丁中氮原子上有孤对电子,是富电子体,可作为电子给予体,两者在溶液中可以形成稳定的1∶1型络合物。基于荷移络合物的组成为1∶1以及2种反应物的结构,推测荷移反应(见图4)。
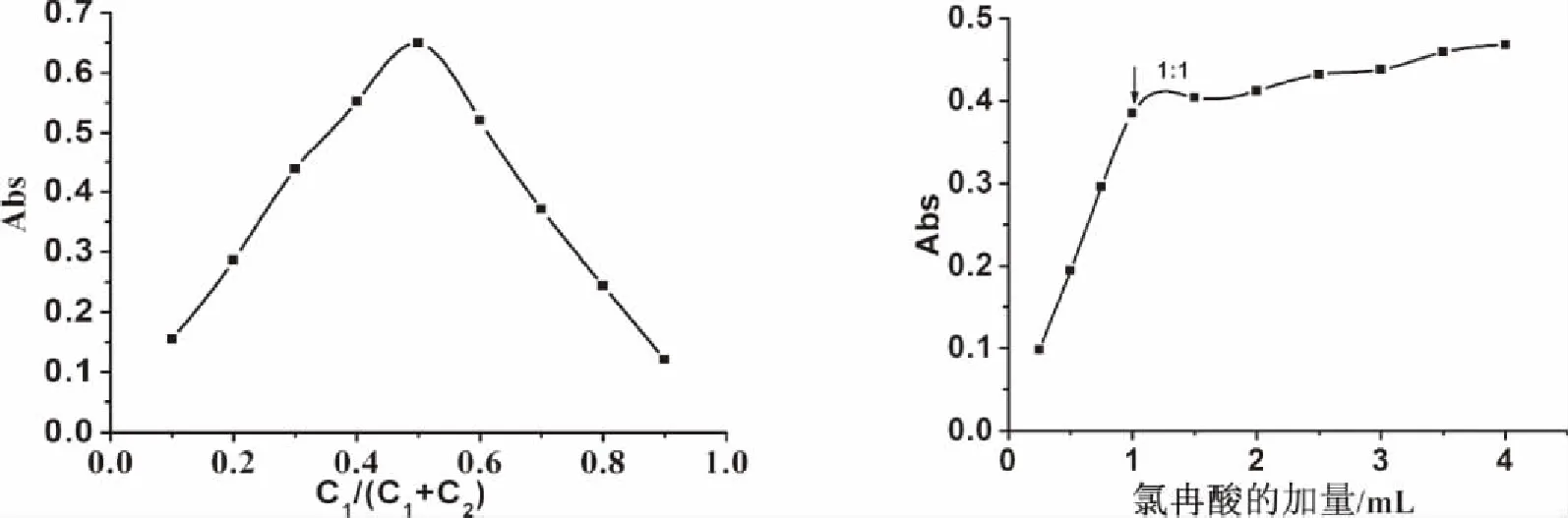
C1:尼扎替丁的初始浓度; C2:氯冉酸的初始浓度。 图2 摩尔连续变化法 图3 摩尔比法

图4 荷移络合物的形成过程
3 讨论
本研究基于氯冉酸电子受体与尼扎替丁电子供体之间的电荷转移反应,建立了一种简单、快速和灵敏的分光光度法测定尼扎替丁制剂中尼扎替丁的含量。对影响反应物形成的因素进行了研究和优化。该方法已成功地应用于测定尼扎替丁分散片和胶囊,所得结果与文献方法得到的结果相吻合。运用等摩尔连续变化法和摩尔比法研究了尼扎替丁与氯冉酸之间的化学计量关系。
电荷转移络合物的形成影响因素有:电子受体和电子供体的结构、溶剂的性质等。电荷转移络合物共振杂化体中有电荷转移的过程,因此增大溶剂的介电常数有利于尼扎替丁-氯冉酸络合物的形成。在本实验中溶剂的介电常数大小是:水>二甲亚砜>乙腈>甲醇>乙醇>1-正丙醇>二氯甲烷,而水和二甲亚砜本身是一个供体分子,不利于尼扎替丁-氯冉酸络合物的形成。虽然乙腈和甲醇的介电常数相差不大,但是尼扎替丁-氯冉酸络合物在乙腈介质中的吸光值远大于在甲醇中的吸光值。所以,本文选择乙腈作为溶剂。电荷转移络合物的生成是一个熵减过程,升温不利于尼扎替丁-氯冉酸络合物的形成,所以本方法选择在室温即可。
段虹飞等[2]用超高效液相色谱串联质谱法快速测定尼扎替丁在人体血浆中的含量,检测限低但是设备较昂贵,检测成本较高,不适合生产中的质量控制;邢海燕等[5]用高效液相色谱法测定尼扎替丁注射液的含量,其中流动相缓冲盐的浓度很高(0.1 mol/L)对液相设备的稳定性影响较大;邱月琴等[6]用荧光探针测定了尼扎替丁的含量,其中灵敏度较高,但相对于本方法的检测时间更长。
基于氯冉酸与尼扎替丁之间的络合反应测定尼扎替丁的含量,该方法简便、快速且成本较低。因此,本法对于尼扎替丁生产中的质量控制有指导意义。
[1] 李焕德.新型H2—受体拮抗剂尼扎替丁[J].国外医学:药学分册,1990,17(4):212-216.
[2] 段虹飞,赵宁民,李巧艳,等.超高效液相色谱串联质谱法快速测定尼扎替丁在人体血浆中的含量[J].中国医院药学杂志,2016,36(5):355-358.
[3] Çakar M B,Ulu S T.HPLC fluorescence method for the determination of nizatidine in human plasma and its application to pharmacokinetic study[J].Luminescence,2014,29(4):357-361.
[4] Zhang L,Wang T.Quantitative determination of nizatidine in human plasma and urine by high performance liquid chromatography and its pharmacokinetic study in humans[J].中国药学:英文版,2010,19(4):279-284.
[5] 邢海燕,杨永.高效液相色谱法测定尼扎替丁注射液的含量[J].现代医药卫生,2009,25(2):203-204.
[6] 邱月琴,常银霞,杜黎明,等.荧光探针测定雷尼替丁、尼扎替丁和西咪替丁[J].药物分析杂志,2012,32(6):973-977.
[7] 易跃能,杨华,叶樱,等.尼扎替丁注射液含量测定的方法比较[J].中南药学,2006,4(1):22-23.
[8] Mulliken R S.Structures of complexes formed by halogen molecules with aromatic and with oxygenated solvents1[J].Journal of the American Chemical Society,1950,72(1):600-608.
[9] Darwish I A,Alshehri J M,Alzoman N Z,et al.Charge-transfer reaction of chloranilic acid with crizotinib:spectrophotometric study,computational modeling and use in development of microwell assay for crizotinib[J].Journal of Solution Chemistry,2014,43(7):1282-1295.
[10]Elqudaby H M,Mohamed G G,Eldin G M.Analytical studies on the charge transfer complexes of loperamide hydrochloride and trimebutine drugs.Spectroscopic and thermal characterization of CT complexes[J].Spectrochimica Acta Part A Molecular & Biomolecular Spectroscopy,2014,129(6):84-95.
猜你喜欢
电子测试(2022年16期)2022-10-17
煤化工(2022年3期)2022-07-08
合成纤维工业(2021年5期)2021-10-31
河北画报(2020年10期)2020-11-26
首都食品与医药(2020年1期)2020-10-21
汽车电器(2019年1期)2019-03-21
花火A(2018年9期)2018-11-26
英美文学研究论丛(2018年2期)2018-08-27
山东工业技术(2016年10期)2016-09-06
雕塑(2000年4期)2000-06-24
