
基于遗传算法的蛋白质复合物识别算法*
2018-05-09 08:50郑文萍李晋玉
计算机与生活 2018年5期
郑文萍,李晋玉,王 杰,2
1.山西大学 计算机与信息技术学院,太原 030006
2.山西大学 计算智能与中文信息处理教育部重点实验室,太原 030006
3.山西大学 大数据挖掘与智能技术山西省协同创新技术中心,太原 030006
1 引言
随着人类基因组计划的实施,生物医学进入后基因时代,系统全面地理解蛋白质之间通过相互作用完成生命活动的规律成为研究热点之一[1]。近年来,随着酵母双杂交、微阵列等高通量技术的应用,产生了大量的蛋白质互作用(protein-protein interaction,PPI)数据,这些数据可以表示为蛋白质互作用网络的形式[2]。通常将蛋白质互作用网络看作一个图G=(V,E),其中节点表示蛋白质,边表示蛋白质之间的相互作用。在蛋白质互作用网络中,蛋白质复合物是指在同一时间和空间组成一个多分子机制的一组蛋白质。从大规模蛋白质互作用网络中识别蛋白质复合物对预测蛋白质功能,解释特定的生物进程具有重要作用。蛋白质复合物往往对应于互作用网络图中的一些稠密子图[3]。基于蛋白质互作用网络的图聚类算法是识别网络中复合物[4-6]的有效方法。
2005年,Palla等人[7]提出了派系过滤算法CPM(clique percolation method),并基于此设计了CFinder(clique finder)[8]工具,寻找网络中所有的k-完全子图(k-团),合并具有k-1个公共节点的k-团形成簇,作为参考蛋白质复合物。2012年,Liu等人[9]对CPM算法进行了改进,提出了基于团渗透和距离限制的蛋白质复合物识别算法CPM-DR(clique percolation method based on distance restriction)。然而,寻找一个图的k-完全子图是NP(non-deterministic polynomial)困难问题。2003年,Bader和Hogue[10]提出了基于局部密度搜索的MCODE(molecular complex detection)算法,以节点邻域的k-核局部网络密度作为PPI网络中节点的权重,并选择权重最大的节点作为种子节点;以种子节点为初始簇中心,迭代向当前簇中加入节点权重大于给定阈值的邻居节点,完成簇扩展过程。MCODE算法时间复杂度为O(n),被广泛应用于大规模网络,然而它不能保证所发现的簇内连接紧密[11]。2006年,Altaf-Ul-Amin等人[12]提出DPClus(development clustering)算法,认为存在相互作用的两个节点之间的公共邻居数越多,则作用越强烈,以此对PPI网络中的边加权,并定义节点权重为加权度和,选择权重最大的节点作为种子节点;DPClus还考虑了簇边界节点对当前簇的影响,给出了节点的簇边界性值,与簇密度结合进行簇扩展,对PPI网络中稠密区域和稀疏区域加以区分;扩展之后形成的稠密区域作为识别的蛋白质复合物。2008年,李敏等人[11]对DPClus的边界性值进行改进,提出了IPCA(improvement development clustering algorithm)算法,将子图直径和子图密度相结合进行簇扩展,并将子图直径作为蛋白质复合物识别特征之一,可以发现重叠的蛋白质复合物,提高了算法效率。2011年,王建新等人[13]给出了对加权蛋白质互作用网络中的功能模块进行识别的层次聚类算法HC-PIN(hierarchical clustering-protein interaction network)。2012年,Nepusz等人[14]综合考虑簇内紧密度和簇间分离性,提出了ClusterOne(clustering with overlapping neighborhood expansion)算法对蛋白质复合物进行识别。
种子节点的选择对于最终形成的簇有很大影响。以上各种计算方法,一旦选定权重最大的节点作为初始簇中心,在簇扩展过程中种子节点不再改变。然而在蛋白质互作用网络中,一方面,具有最大权重的节点往往不是唯一的;另一方面,尽管被选作簇中心的节点通常权重较大,但仅选择最大权重的节点作为种子节点进行扩展,在一定程度上缩小了簇发现过程的搜索空间。此外,在簇扩展过程中,一个节点一旦被加入到某一个簇中,再不会被调整出簇,这种方法也缩小了簇发现的搜索空间。
以启发式为代表的智能优化算法可以通过种群进化的方式提高簇发现过程的多样性,目前已经逐渐发展为一种有竞争力的复合物发现方法。在文献[15-17]中,蚁群算法被用来进行社区发现和蛋白质功能模块识别。遗传算法作为智能优化算法的一种,近年来已经被用来与聚类算法相融合,进行蛋白质复合物识别。2012年,Mukhopadhyay等人[18]提出了基于多目标进化的复合物识别算法PROCOMOSS(protein complex detection using multi-objective evolutionary approach based on semantic similarity),个体表示复合物(簇),用k-means算法产生初始种群,为了得到内部连通的子网络作为簇,算法仅采用变异方式使种群进化,没有使用交叉操作。2015年,Emad等人[19]用遗传算法对蛋白质互作用网络进行复合物识别,个体表示一种簇划分结果,同样也仅通过变异操作实现种群进化。2015年,Cao等人[20]提出了一种算法MOEPGA(multi-objective evolutionary programming genetic algorithm),在进化过程中抽取非支配子图作为预测簇,通过对两个有公共节点的子图添加边的方式产生变异,调整簇的结构。
以上基于种群进化方法在PPI网络中进行蛋白质复合物发现的算法中,直接进行交叉操作会产生大量的内部不连通的簇,因此大多仅采用了变异的种群进化方式。本文提出一种基于遗传算法的蛋白质复合物识别的图聚类算法GAGC(genetic algorithm based graph clustering),其中个体表示一种聚类结果(类别之间可能存在重叠节点)。首先对IPCA算法[11]进行改进产生初始种群;然后设计了基于网络对齐的交叉进化方式产生下一代种群;以F-measure值作为种群进化的目标函数。与DPClus、MCODE、IPCA、ClusterOne、HC-PIN、CFinder算法进行了对比实验,结果表明GAGC算法能够扩大图聚类算法的搜索空间,提高解的多样性,进而提高蛋白质复合物检测的性能。
2 基本概念
蛋白质互作用网络可以表示为一个简单无向图G=(V,E),其中节点集合V表示蛋白质集合,边集E表示蛋白质之间相互作用集合。
节点v的邻域Nv={u|(u,v)∈E},定义节点v对子图K(v∉V(K))的连接概率为:

子图K直径D(K)表示子图K中任意一对节点之间最短路径的最大长度,子图K的一阶邻域定义为
图G的顶点子集S的导出子图G[S]是由顶点集S和G中连接S中节点的边所构成的子图,即V(G[S])=S,E(G[S])={(u,v)|(u,v)∈E,u∈S,v∈S}。在不引起混淆的情况下,简记G[S]为S。
本文采用重叠分数(overlapping score,OS)[21]衡量两个子图G1和G2的相似性,定义见式(2)。显然,两个子图具有的公共节点越多,重叠分数值越高。
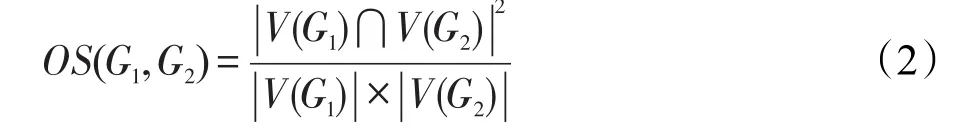
如果G1和G2表示两个蛋白质复合物,则式(2)可以用来衡量这两个复合物组成成分的相似性,其中V(Gi)表示组成复合物Gi的蛋白质集合。两个复合物拥有的公共蛋白质越多,则它们越相似。通常会设定一个阈值ω,如果OS(G1,G2)≥ω,则两个复合物是相匹配的。在蛋白质复合物识别问题中通常取ω=0.2[9-13,22]。
3 基于遗传算法的蛋白质复合物识别算法GAGC
在蛋白质互作用(PPI)网络中进行复合物识别,本质上是对PPI网络所对应的图G进行图聚类,最终所得的一个簇对应一种蛋白质复合物。图聚类作为一种NP困难的组合优化方法,使用启发式搜索方法可以保证在允许的时间内给出满意的图聚类结果,它的两个关键环节是种子节点的选择过程和簇扩展过程。遗传算法(genetic algorithm,GA)是一种有效的全局优化计算技术,采用群体搜索对当前种群采用选择、交叉、变异等基本操作,产生新一代种群,通过种群进化达到或接近最优解。本文设计了一种基于遗传算法的图聚类方法GAGC在蛋白质互作用网络中进行复合物发现。文中一条染色体(个体)对应一个可行的图聚类结果,染色体由多个相互之间有重叠的基因组成,每个基因对应一个可能的蛋白质复合物,算法主要包括初始种群优化、选择、交叉、变异等基本过程。
3.1 目标函数-个体适应度
适应度是遗传算法的重要指标,用来衡量种群中个体的优劣程度,其函数值大小影响到种群中每个个体的生存几率的大小,对算法的性能和收敛速度有很大影响。GAGC选用F-measure作为个体的适应度评价函数,其综合了精准率(Precision)和召回率(Recall)两个指标。精准率是算法正确识别的复合物数与算法得到的复合物总数之比,召回率是标准数据库中蛋白质复合物被识别的准确率,如式(3)~(7)所示。其中,重叠分数(OS)[21]计算复合物的匹配情况。通常,对于复合物P1和P2,如果OS(P1,P2)≥0.2,则可以认为两个复合物是相匹配的[9-13,22]。
令P表示算法得到的蛋白质复合物集合,B表示标准数据集中蛋白质复合物的集合。定义:
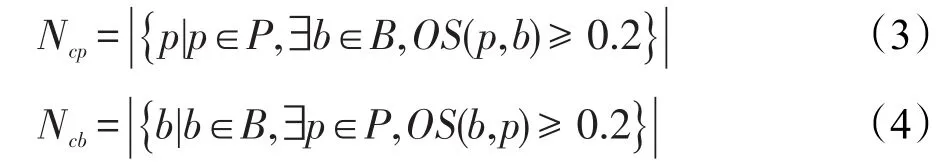
利用式(3),定义算法精准率如式(5)所示。

利用式(4),定义算法召回率如式(6)所示。

根据式(5)、(6),可以得到某个体的F-measure值。
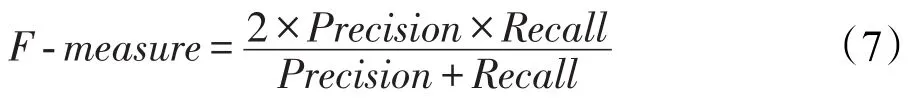
本文采用F-measure值作为算法GAGC的适应度函数来衡量个体的优劣。
3.2 初始种群产生过程
本文采用算法IPCA[11]产生一组可行的图聚类结果作为初始种群。IPCA算法首先对网络的节点权重进行定义,选择权重最大的节点作为种子节点并扩展成为簇。仅选择最大权重节点作为种子节点进行簇扩展,无法保证初始种群的多样性。因此,本文对IPCA算法进行改进,选择种子节点的过程中加入轮盘赌机制,一个节点权重越大,成为种子节点的概率越大。这样,既可以保证选择的种子节点在概率意义下有较大权重,也可以确保种子节点选择的多样性,并最终扩展得到图聚类的多个可行解作为初始种群。
初始种群的产生过程如算法1所示。
算法1初始种群构造子过程InitialPopulation
输入:图G=(V,E),参数Tin和直径参数d,种群大小k(默认k=50)。
输出:初始种群P={P1,P2,…,Pk}。
步骤1令t=1,根据式(8)和式(9)分别计算G中边权重EW(u,v)和顶点权重NW(u),并将顶点按权重值非递增排序。


步骤2令候选种子节点集合HS=V,Pt=∅。
步骤3若HS非空,转步骤4;若节点vi∈HS,则其被选择成为种子节点的概率是
步骤4按照轮盘赌机制选择HS中的一个节点v作为种子节点,令K={v},HS=HS-{v}。
步骤5在子图K的一阶邻域NK中,如果存在节点u满足IN(u,K)>Tin且D(G[K⋃{v}])≤d,则令K=K⋃{u}和HS=HS-{u},转步骤5;若不存在节点u,令pt=pt⋃{K},转步骤3。
步骤6t=t+1,如果t≤k,转步骤2;否则,结束并输出初始种群P。
3.3 选择策略
为了体现遗传算法适者生存的思想,使得优质个体参与进化,从当前种群中选择个体去产生下一代个体,此处选择适应度值较高的个体进行进化并成为下一代种群中的个体。如果单纯按照适应度值从高到低顺序选择,会降低种群的多样性,使得遗传算法早熟,因此此处采用轮盘赌方式,选择成为下一代个体的概率与节点的适应度成正比。设种群大小为n,每个个体的适应度为fi,则i被选中的概率为。这样优秀个体会以更大概率被选择,选择的方法是:
(3)在[0,1]之间产生一个随机数rand,若rand≤prob1,则个体1被选中,若probi-1<rand≤probi,则选择第i个个体。
重复以上操作n0次,则产生了n0个个体构成的新的种群,本文取初始种群规模n0=50。
除此之外,为了保留每一代种群中的最优个体,在产生了新的用于进化下一代的种群后,如果当前种群中的最优个体的适应度值低于上一代最优个体的适应度值,则使用上一代中的最优个体替换当前所选种群中的最差个体。
3.4 基于染色体对齐的交叉策略
交叉操作是遗传算法中最重要也是最基本的遗传操作。本文算法中,如果直接选择两个染色体进行交叉,可能使得交叉结果中存在大量节点没有簇归属,影响聚类效果。为了更有效地进行交叉操作,本文算法首先根据重叠分数将两个染色体中的基因对齐,使两个等位基因表示的子图有比较多的重复节点。交叉操作应该在等位基因上进行,这样可以尽可能保证基因交叉后产生的新子图是连通的。
3.4.1 染色体对齐机制
GAGC算法中的一个染色体(个体)代表一个图聚类结果,染色体的每个基因代表一个簇,即一个顶点子集。此处采用式(2)定义的重叠分数OS作为两条染色体对齐的依据。
设S=(s1s2…sx)和R=(r1r2…ry)是两个长度分别为x和y的染色体。首先构造S和R的相似矩阵Mx×y,其元素mi,j=OS(si,rj)。
不失一般性,假设x≥y,具体算法描述如下。
染色体对齐算法Alignment(S,R,x,y):
1.ints1[x],r1[y];//初始化为0
2.For(i=1;i<=x;i++)
3.{
4.intmaxi=0;
5.For(j=1;j<=y;j++)
6.{k=0;
7. if(OS(s[i],r[j])>maxi&&r1[j]==0)
8. {maxi=OS(s[i],r[j]);k=j;}
9.}
10.if(maxi>=0.8){s1[i]=k;r1[k]=1;}
11.}
12.For(j=1;j<=x;j++)
13.if(s1[j]!=0)tempr[j]=r[s1[j]];
14.For(j=1;j<=y;j++)
15.r[j]=tempr[j];
选择两条染色体中长度较长的染色体(假设为染色体s)作为参照染色体,对s的每一个基因si,在染色体r中选择未被使用过的OS(si,rj)≥0.8且重叠分数最大的基因作为si的等位基因。如果等位基因不存在,则用空位表示。
对图1(1)中的染色体1和染色体2执行对齐操作的过程如下。

首先,根据重叠分数定义,构建两个染色体的相似矩阵M:然后,以较长的染色体1为参照,进行对齐。对染色体1的每个基因pi,遍历相似矩阵,得到与该基因重叠分数值最高且尚未对齐的染色体2中的基因q,称q为pi的等位基因。将q在染色体2中移动到与pi对应的位置。
如果染色体1中某个基因无法在染色体2中找到等位基因,则将其移动到染色体1的末尾位置。
图1(2)给出了两个染色体对齐后的结果图。
3.4.2 交叉策略
遗传算法的交叉机制是优化的关键环节。为了扩大可行解的搜索空间,增加个体多样性,GAGC算法主要通过交叉操作改变染色体上的等位基因(复合物)。本文选用传统遗传算法的单点交叉机制,交叉时每次都以种群中具有最高适应度的个体作为父本,从剩余的个体中随机选择母本与其进行交叉。交叉方式为:首先将选择的两个个体染色体对齐,然后随机产生一个交叉位,个体染色体交叉位前后互换染色体片段,即等位基因互换。如图2所示,来自父本的染色体和来自母本的染色体交叉时,产生的交叉点为染色体的位置1,此时将父本与母本的染色体交叉位置之后的染色体片段互换,形成两个新的子代。
交叉算法过程如算法2所示。
算法2交叉子过程GAGC-CROSSOVER
输入:父本染色体F和母本染色体M。
输出:新的染色体son1、son2。
步骤1按照轮盘赌方式选择当前种群个体适应度最高的个体作为父本F,再从当代种群中随机选择另外一个个体作为母本M。
步骤2对M和F执行染色体对齐算法Alignment。
步骤3随机产生一个交叉点T;交换两条染色体T位置之前和之后的基因(复合物)。
图2给出了父本和母本交叉操作示意图。
3.5 变异策略

Fig.1 Schematic diagram of chromosome alignment图1 染色体对齐示意图
交叉操作产生的子代个体除了继承父代个体的信息外,还会按一定的概率发生变异,这体现了生物遗传的多样性。变异操作是以固定概率Pm进行的。变异时,首先随机产生一个变异位,对应于该染色体上的一个基因(簇)C。随机选择该复合物中的一个蛋白质g,考虑g在对应互作用网络中的邻域Ng。若Ng-C≠∅,则随机选择节点v∈Ng-C,将v加入该复合物中;如果Ng-C=∅,则删除g。图3给出了某蛋白质的变异过程示意图。

Fig.2 Schematic diagram of crossover operation图2 交叉操作示意图

Fig.3 Schematic diagram of variation operation图3 变异操作示意图
3.6 基于遗传算法的图聚类算法GAGC
GAGC算法的流程如下所示。
算法3基于遗传算法的蛋白质复合物识别图聚类算法GAGC
输入:G=(V,E),种群规模n0=50,k=100,Pm=0.3(变异率),最大迭代次数y=50。
输出:复合物集合C。
令t=0;
步骤1使用算法InitialPopulation产生100个初始个体。
步骤2执行3.3节所述的选择策略,选择n0个个体作为当前种群。
步骤3在当前种群中,执行n0次交叉操作GAGC-GROSSCOVER,产生2n0个子代个体。
步骤4对每个子代个体,随机产生(0,1)的数Rf,如果Rf<Pm,则该个体执行变异操作。
步骤5t=t+1,若t<y,则转步骤2。
步骤6输出当前种群最优个体C作为最终聚类结果。
4 实验结果与分析
4.1 实验数据和评价标准
本文互作用网络数据来自于DIP[23]和GAVIN[24]两个酿酒酵母蛋白质互作用网络,在移除自环和重复的互作用后,DIP网络最终包含4 930个节点和17 201条边,GAVIN数据集包括1 430个节点和6 531条边。采用的复合物标准集为CYC2008[25]标准集,其中包含由1 628个蛋白质构成的408个蛋白质复合物。
为了评估算法检测出的蛋白质的性能,本文采用重叠分数OS、精准率、召回率和F-measure指标进行算法评价。其中重叠分数OS是用来衡量两个复合物的匹配效果的度量方式。以上4种评价方式在个体适应度计算模块中均已介绍。除此之外,选用Accuracy作为算法的评价指标。Accuracy由敏感度(Sn)和真阳性预测值(PPV)构成。敏感度(Sn)表示识别的在标准复合物中覆盖蛋白质的多少,真阳性预测值(PPV)表示识别的蛋白质复合物成为真阳性的可能性。其定义如下:

其中,n表示标准集中复合物的个数;m表示算法识别的复合物个数;Tij表示第i个标准复合物与第j个算法所识别的复合物之间公共蛋白质数目。
4.2 实验结果
为了分析加入遗传算法的GAGC与IPCA的效果。本文分别比较了两个算法在Tin取值0.1至0.9时F-measure和Precision的变化曲线图。从图4和图5中可知,两个算法Tin的变化趋势大致相同,在Tin为0.5时,二者的F-measure均达到最大值,在Tin为0.4时,二者的Precision均达到最大值,但GAGC在不同参数的Precision和F-measure值均高于IPCA的取值,并且在F-measure值上提升近5%,在Precision值上提升近10%。这也就表明了加入遗传算法对IPCA的改进效果是明显的。在GAVIN数据上F-measure值和Precision如图4和图5所示。
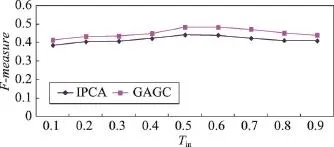
Fig.4 F-measurevalues of GAGC and IPCAon GAVIN data图4GAGC与IPCA在GAVIN数据上的F-measure值
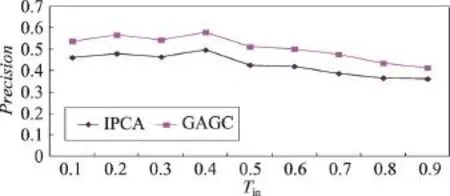
Fig.5 Precisionvalues of GAGC and IPCAon GAVIN data图5GAGC与IPCA在GAVIN数据上的Precision值

Table 1 Performance comparison of different methods with GAGC on GAVIN data表1 几种算法与GAGC算法在GAVIN数据上的对比分析
为了分析GAGC算法的性能,本文对比了DPClus、MCODE、IPCA、HC-PIN、ClusterOne、CFinder算法在DIP数据和GAVIN数据上的8个指标Ncb、Ncp、Precision、Recall、F-measure、Sn、PPV、Accuracy的值。这里GAGC和IPCA的参数Tin=0.5,其他算法的参数根据作者给定的最优参数值。由于遗传算法是一种随机算法,这里GAGC在相同参数下重复进行30次实验,计算每个指标的均值与标准差,与其他算法进行对比,结果如表1和表2所示。从表1、表2可知GAGC在Recall上与IPCA相同,均为最高,并且在F-measure值上GAGC高于IPCA、DPClus、MCODE、CFinder、HC-PIN、ClusterOne算法,但精准率相对较低。其主要原因为GAGC算法在发现蛋白质复合物时并未进行后期的处理,即将所聚类的簇都看作是蛋白质复合物,这样就会导致算法产生的蛋白质复合物数目偏大,从而精确率会低于其他算法。并且在其他3个指标上,GAGC也与其他6种算法效果相当。

Table 2 Performance comparison of different methods with GAGC on DIP data表2 几种算法与GAGC算法在DIP数据上的对比分析
5 结束语
本文提出了一种基于遗传算法的蛋白质复合物识别算法GAGC。为了扩大算法的搜索空间,首先改进了IPCA算法,产生初始种群;为了能够将遗传算法的交叉操作运用到复合物识别中,设计了基于重叠分数OS的染色体对齐方式;然后通过设计交叉方式,扩大簇形成的搜索空间,从而提高算法识别蛋白质复合物的精度。该算法框架也可以扩展到其他聚类算法,通过选择合适的适应度函数,提升聚类算法的性能。
[1]Garrels J I.Yeast genomic databases and the challenge of the post-genomic era[J].Functional&Integrative Genomics,2002,2(4):212-237.
[2]Guo Maozu,Dai Qiguo,Xu Liqiu,et al.On protein complexes identifying algorithm based on the novel modularity function[J].Journal of Computer Research and Development,2014,51(10):2178-2186.
[3]Spirin V,Mirny LA.Protein complexes and functional modules in molecular networks[J].Proceedings of the National Academy of Sciences,2003,100(21):12123-12128.
[4]Yu Liang,Gao Lin,Sun Penggang.Research on algorithms for complexes and functional modules prediction in proteinprotein interaction networks[J].Chinese Journal of Computers,2011,34(7):1239-1251.
[5]Li Xueyong,Wang Jianxin,Zhao Bihai,et al.Identification of protein complexes from multi-relationship protein interaction networks[J].Human Genomics,2016,10(S2):61-70.
[6]Li Min,Wu Xuehong,Wang Jianxin,et al.Towards the identification of protein complexes and functional modules by integrating PPI network and gene expression data[J].BMC Bioinformatics,2012,13(1):1-15.
[7]Palla G,Derényi I,Farkas I,et al.Uncovering the overlapping community structure of complex networks in nature and society[J].Nature,2005,435(7043):814-818.
[8]Adamcsek B,Palla G,Farkas I J,et al.CFinder:locating cliques and overlapping modules in biological networks[J].Bioinformatics,2006,22(8):1021-1023.
[9]Liu Binbin,Li Min,Wang Jianxin,et al.An algorithm for identifying protein complexes based on clique percolation and distance restriction[J].System Engineering-Theory and Practice,2012,32(2):389-397.
[10]Bader G D,Hogue C W V.An automated method for finding molecular complexes in large protein interaction networks[J].BMC Bioinformatics,2003,4(2):20-29.
[11]Li Min,Chen Jian'er,Wang Jianxin,et al.Modifying the DPClus algorithm for identifying protein complexes based on new topological structures[J].BMC Bioinformatics,2008,9(1):1-16.
[12]Altaf-Ul-Amin M,Shinbo Y,Mihara K,et al.Development and implementation of an algorithm for detection of protein complexes in large interaction networks[J].BMC Bioinformatics,2006,7(1):207-219.
[13]Wang Jianxin,Li Min,Chen Jian'er,et al.A fast hierarchical clustering algorithm for functional modules discovery in protein interaction networks[J].IEEE/ACM Transactions on Com-putational Biology&Bioinformatics,2011,8(3):607-620.
[14]Nepusz T,Yu Haiyuan,Paccanaro A.Detecting overlapping protein complexes in protein-protein interaction networks[J].Nature Methods,2012,9(5):471-476.
[15]Liu Yan,Wang Qingxian,Wang Qiang,et al.Email community detection using artificial ant colony clustering[C]//LNCS 4537:Proceedings of the 2007 International Workshops Advances in Web and Network Technologies,and Information Management,Huangshan,Jun 16-18,2007.Berlin,Heidelberg:Springer,2007:287-298.
[16]Ji Junzhong,Liu Zhijun,Zhang Aidong,et al.HAM-FMD:mining functional modules in protein-protein interaction networks using ant colony optimization and multi-agent evolution[J].Neurocomputing,2013,121(18):453-469.
[17]Chang Honghao,Feng Zuren,Ren Zhigang.Community detection using ant colony optimization[C]//Proceedings of the 2013 IEEE Congress on Evolutionary Computation,Cancun,Jun 20-23,2013.Piscataway:IEEE,2013:3072-3078.
[18]Mukhopadhyay A,Ray S,De M.Detecting protein complexes in a PPI network:a gene ontology based multi-objective evolutionary approach[J].Molecular Biosystems,2012,8(11):3036-3048.
[19]Emad R,Ahmed N,Moataz A.Protein complexes predictions within protein interaction networks using genetic algorithms[J].BMC Bioinformatics,2016,17(S7):481-543.
[20]Cao Buwen,Luo Jiawei,Liang Cheng,et al.MOEPGA:a novel method to detect protein complexes in yeast proteinprotein interaction networks based on multiobjective evolutionary programming genetic algorithm[J].Computational Biology&Chemistry,2015,58:173-181.
[21]Brohée S,Helden J V.Evaluation of clustering algorithms for protein-protein interaction networks[J].BMC Bioinformatics,2006,7(1):1-19.
[22]Wang Jianxin,Peng Xiaoqing,Li Min,et al.Construction and application of dynamic protein interaction network based on time course gene expression data[J].Proteomics,2013,13(2):301-312.
[23]Xenarios I,Salwínski L,Duan X J,et al.DIP,the database of interacting proteins:a research tool for studying cellular networks of protein interactions[J].Nucleic Acids Research,2002,30(1):303-305.
[24]Gavin A C,Aloy P,Grandi P,et al.Proteome survey reveals modularity of the yeast cell machinery[J].Nature,2006,440(7084):631-636.
[25]Pu Shuye,Wong J,Turner B,et al.Up-to-date catalogues of yeast protein complexes[J].Nucleic Acids Research,2009,37(3):825-831.
附中文参考文献:
[2]郭茂祖,代启国,徐立秋,等.一种蛋白质复合体模块度函数及其识别算法[J].计算机研究与发展,2014,51(10):2178-2186.
[4]鱼亮,高琳,孙鹏岗.蛋白质网络中复合体和功能模块预测算法研究[J].计算机学报,2011,34(7):1239-1251.
[9]刘彬彬,李敏,王建新,等.基于团渗透和距离限制的蛋白质复合物识别算法[J].系统工程理论与实践,2012,32(2):389-397.
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
今日农业(2021年12期)2021-11-28
西华大学学报(自然科学版)(2020年6期)2020-10-15
上海理工大学学报(2020年4期)2020-09-27
初中生世界·八年级(2019年6期)2019-08-13
计算机与数字工程(2019年3期)2019-03-26
安徽化工(2018年4期)2018-09-03
计算机应用(2018年7期)2018-08-27
小学生导刊(低年级)(2016年9期)2016-10-13
小学生导刊(低年级)(2016年6期)2016-07-02
