
3-环丙基-1-甲基-1H-吡唑-4-甲醛的合成研究
2018-09-04 06:17王先恒赵长阔周亦琪
浙江化工 2018年8期
王先恒,赵长阔,袁 智,曹 颖,周亦琪
(遵义医学院 药学院,贵州 遵义 563099)
吡唑类 (pyrazole)杂环化合物是一类包含1,2-二氮的五元芳杂环的重要医药中间体,很多包含此类结构的化合物都显示出抗真菌、抗病毒、抗肿瘤、杀虫等多种多样生物活性[1-4];该类中间体一直是药物学家关注的热点。而环丙基是一个重要的药效基团,很多药物由于环丙基的引入而药效增强,如环丙沙星等。
研究发现,3-环丙基-1-甲基-1H-吡唑-4-甲醛是合成一些吡唑稠合环以及包含4-吡唑醛的活性先导物质的关键中间体,如图1所示的化合物 a[5]、b[6]、c[7]。 例 如 ,Merck 制药 在 PCT 专利WO2015090507A1中公开了四氢四唑并[1,5-a]吡嗪化合物,该类化合物可抑制类维生素A相关的孤儿受体ROR-γ(Retinoid-related orphan receptorγ),用于治疗受RORγ调节的类风湿性关节炎、多发性硬化症、牛皮癣、溃疡性结肠炎、哮喘、自身免疫性肝炎或1型和2型糖尿病的医学病症[5]。辉瑞制药在PCT专利WO2012137089A1中公开了原肌球蛋白相关激酶Trks(Tropomyosin-related kinases),该类激酶通过神经营养因子激活,在疼痛感及肿瘤细胞生长和存活信号中起重要作用,抑制Trks激酶可能为诸如疼痛和癌症等病症提供靶向治疗[6]。Bavetsias V公开了咪唑并[4,5-b]吡啶类,是一种双 FLT3/极光激酶 (FLT3/Aurora Kinase)抑制剂,用于治疗急性髓细胞性白血病[7]。

图1 包含3-环丙基-1-甲基-吡唑醛中间体的活性结构
3-环丙基-1-甲基-1H-吡唑-4-甲醛是合成上述活性先导物质 (化合物a、b、c)的关键中间体,目前还没有文献直接公开该化合物的制备。
路莹莹等[8]公开了1H-吡唑-4-甲醛的合成方法,以氰基乙酸乙酯为起始原料,通过用原甲酸酯进行α-甲酰化、与水合肼缩合后形成吡唑环、重氮化脱除氨基、还原酯键得到醇,最后经MnO2氧化得到相应的醛,共五步反应得到目标化合物1H-吡唑-4-甲醛(1)。然而该方法不仅路线比较长,还用到了锂铝氢等较为昂贵的试剂;此外,重氮化反应也需要小心操作。

图2 1H-吡唑-4-甲醛1的合成路线[7]
本文通过查阅大量文献,设计了一条操作简便、原料价廉易得且收率较高的合成路线。
1 材料与仪器
1.1 材料
1-环丙基甲基酮、二甲氧基-N,N-二甲基甲胺、盐酸肼、硫酸二甲酯 (分析纯,Aldrich)、β-氯丙酰氯、三乙基硅烷、浓盐酸 (37%,国药集团化学试剂有限公司);无水硫酸钠、无水硫酸镁、碳酸氢钠、碳酸钠、乙酸钠、三氯氧磷 (化学纯,国药集团);二氯甲烷、氯仿、N,N-二甲基甲酰胺、甲醇、乙醇、乙酸乙酯、石油醚、己烷(化学纯,成都市科龙化工试剂厂);硅胶薄层板(青岛海洋化工厂)、柱层析用硅胶 (200~300目);自制纯化水。
1.2 仪器
电热恒温鼓风干燥箱(上海精宏实验设备有限公司,DMG-914M型);暗箱三用紫外分析仪(上海司乐仪器有限公司,81-2型);Varian 400 MHz核磁共振波谱仪(TMS作内标,安捷伦科技有限公司);LC-10ATVP型高效液相色谱仪(日本岛津公司)。
2 合成方法与讨论
2.1 化合物1的合成路线
以1-环丙基甲基酮和二甲氧基-N,N-二甲基甲胺为起始原料,依次通过羟醛缩合、与肼缩合后形成吡唑环、选择性甲基化反应、最后用三氯氧磷与DMF通过Vilsmeier-Hack反应引入醛基,得目标产物3-环丙基-1-甲基-1H-吡唑-4-甲醛 (化合物1),总收率高达48.0%。
2.1.1 (E)-1-环丙基-3-(二甲基氨基)丙-2-烯-1-酮(2)的制备
将 1-环丙基甲基酮(28.57 g,34 mmol)和二甲氧基-N,N-二甲基甲胺 (DMF-DMA,60.71 g,510 mmol)置于圆底烧瓶中,于室温下搅拌2 h,再将混合物加热至120℃搅拌1天。将反应溶液真空浓缩至干,将残余物用二氯甲烷萃取后,用水洗涤并用无水硫酸镁干燥。减压旋转蒸发去除二氯甲烷后,将得到的粗品用冷的己烷洗涤并干燥得到目标化合物1 (40.07 g,85%)。1H NMR(400 MHz, CDCl3)δ=0.73~0.77 (m, 2H), 0.99~1.05 (m, 2H), 1.76~1.82(m, 1H), 2.93(s, 6H),5.20~5.24 (d, J=16 Hz, 1H), 7.56~7.60 (d, J=16 Hz,1H)。
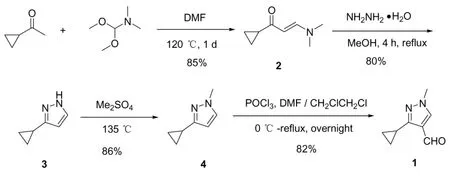
图3 化合物1的合成路线
2.1.2 3-环丙基-1H-吡唑(3)的制备
在冰浴中,向化合物 2(5.00 g,3.6 mmol)的甲醇 (30 mL) 溶液中加入NH2NH2·2HCl的水溶液(4.15 g,4.0 mmol,溶于 30 mL 水中)。 将得到的混合物加热回流2 h,冷却至室温,然后用无水NaHCO3中和。旋转蒸发去除甲醇后,剩余物用二氯甲烷萃取 (30 mL×3)后,合并有机相并用无水硫酸镁干燥。除去有机溶剂,得到目标化合物2(3.11 g,80% ).1H NMR (400 MHz,CDCl3)δ =0.70~0.76 (m, 2H), 0.91~0.95 (m, 2H), 1.91~1.97(m, 1H), 5.97~5.98 (d, 1H), 7.47~7.48 (d,J=16 Hz, 1H), 7.94 (s, 1H).
2.1.3 3-环丙基-1-甲基-1H-吡唑(4)的制备
将硫酸二甲酯(7.8 g,61.7 mmol)置于圆底烧瓶中,于室温下逐滴加入到上述制备得到化合物3(3.3 g,30.8 mmol),加料完毕后将混合物加热至135℃继续反应4 h。将反应液冷却至室温,加入饱和碳酸钠溶液淬灭反应,用二氯甲烷萃取并用无水硫酸钠干燥。旋转蒸发除去有机溶剂后,将所得粗产物通过柱层析纯化 (用EtOAc:PE=1∶15洗脱),得到化合物 3 (3.26 g,86%)。1H NMR(400 MHz, CDCl3)δ=0.69~0.74 (m, 2H), 0.86~0.93(m, 2H), 1.91~1.96(m, 1H), 3.83 (s, 3H),5.89 (s, 1H),7.21 (s,1H).
2.1.4 3-环丙基-1-甲基-1H-吡唑-4-甲醛(1)的制备
在冰水浴中将三氯氧磷 POCl3(10.0 g,65 mmol)滴加至 DMF(5.36 g,7.32 mmol),将该混合物在室温下搅拌2 h。向混合物中加入20 mL二氯乙烷,在冰水浴中逐滴加入化合物4(4.43 g,36 mmol)的15 mL二氯乙烷溶液。加料完毕后,混合物升温至室温,并继续加热至回流温度下搅拌反应过夜。混合物冷却至室温,缓慢加入乙酸钠(16.3 g,0.2 mol)的 50 mL 水溶液。所得的混合物再次回流1 h,随后冷却至室温。混合物在分液漏斗中分层,收集有机层;水层用乙醚萃取。合并有机层用无水硫酸镁干燥,真空浓缩后得到粗产品。粗品通过硅胶柱层析纯化(用EtOAc:PE=1∶5洗脱),得到目标化合物 1 (4.43 g,80%)。1H NMR (400 MHz, CDCl3) δ=0.98~1.01 (m, 4H),2.30~2.38 (m, 1H), 3.83(s, 3H), 7.75 (s, 1H),9.95 (s, 1H)。LC-MS 151.1(MH+);HPLC purity:99.5%。
2.2 结果与讨论
2.2.1 羟醛缩合反应

我们采用活泼性DMF-DMA代替反应性较差的DMF与环丙基甲基酮发生羟醛缩合,得到目标产物2。通过核磁共振谱图的耦合常数J=16 Hz,判断该缩合产物为反式构型。
2.2.2 甲基化反应
一般来说,对吡唑进行烷基化反应,都会有异构体生成。然而,我们发现,3-环丙基-1H-吡唑的烷基化反应,只有一个主要产物4生成,而没有异构体4’生成,分离收率达到86%,具有非常高的选择性。这可能是由于当吡唑氮原子的邻位有大的取代基时,由于空间位置的作用,邻位取代会非常困难,因而生成的产物主要以间位为主,从而反应具有很高的选择性。

3 结论
本文发展了一条以环丙基乙酮和二甲氧基-N,N-二甲基甲胺为起始原料,依次通过羟醛缩合、与肼缩合成环、选择性甲基化反应和Vilsmeier-Hack反应得到目标产物3-环丙基-1-甲基-1H-吡唑-4-甲醛 (化合物1),总收率高达48.0%。该方法为3-环丙基-1-甲基-1H-吡唑类衍生物的合成提供了一条新的选择。
猜你喜欢
化学工程师(2022年3期)2022-04-19
今日农业(2021年2期)2021-11-27
上海化工(2021年2期)2021-04-23
今日农业(2020年23期)2020-12-31
含能材料(2020年7期)2020-07-11
中成药(2018年6期)2018-07-11
中国有色金属学报(2018年2期)2018-03-26
中国医药指南(2017年3期)2017-11-13
中成药(2017年5期)2017-06-13
山西青年(2017年2期)2017-01-11
