
Aln(n=2~10)团簇结构和性质 的密度泛函理论研究
2018-10-22 04:02李东明温俊青陈海霞
西北师范大学学报(自然科学版) 2018年5期
李东明,温俊青,陈海霞
(1.西安石油大学理学院,陕西西安 710065; 2.陕西师范大学物理与技术信息学院,陕西西安 710119)
过渡金属团簇在高密度磁记录等领域具有潜在应用价值[1-2].Aln团簇的电子结构简单,在中等尺寸时就表现出了块体结构的生长趋势,当尺寸减小到几个或者几十个原子时,结构和性质会发生明显的变化.在1986年,Cox等[3]观察到了铝团簇的磁现象,得出每个铝团簇Aln(n≤9)有一个磁矩的结论.自此,对铝团簇的几何与电子结构的研究得到了一系列进展[4-12].众所周知,Aln(n≤5)是平面结构,Aln(n≤11)是在基于Al6的八面体结构的基础上增加原子而成.Akola等发现Aln(12≤n≤23)团簇的生长规律是在Al13的二十面体结构基础上通过加减原子堆积而成[13].Chuang 等[9]发现Al19最稳定结构是在二十面体上加5个邻近的原子和单原子堆积而成,与前人的研究结果为双二十面体不同.Akola等[13-16]研究了负离子铝团簇的结构,发现负离子和中性铝团簇的结构是一致的.Rao等[8]分析了Aln(1≤n≤15)团簇的磁性,表明当n≤10时,奇数个原子的平均原子磁矩是1μB,偶数个原子的平均原子磁矩为2μB,当n≥10时,团簇的磁矩消失,与实验结果相吻合.探索含铝团簇的结构规则,进一步寻找特殊稳定铝化合物团簇,对理解现有材料的结构和性质具有重要的意义.
文中在密度泛函理论中的BPW91/LANL2DZ水平下系统优化了Aln(n=2~10)团簇的结构,分析了团簇的稳定性、磁性随团簇尺寸的演化,并与已有的理论和实验结果进行了比较.所有计算均在 Gaussian 03程序下进行.
1 计算方法
为了确定所选方法的可靠性,选用不同交换关联泛函,基组为LANL2DZ[17],计算了Al2的键长和结合能,并与实验结果进行了比较(表1).
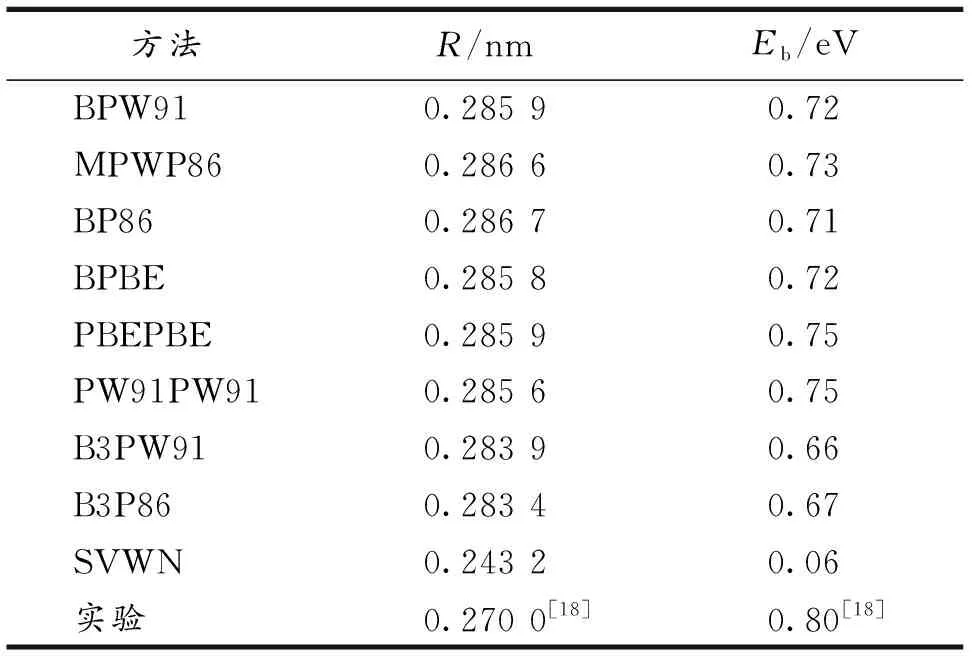
表1 不同交换相关泛函下优化的Al2团簇的键长R(nm), 结合能Eb/(eV)的理论值和实验值
对Al2实验得到基态为三重态,键长为0.27 nm,结合能为0.80 eV[18].从表1可以看到,局域密度近似泛函SVWN方法得到的键长、结合能与实验值相差较远.杂化密度泛函得到的键长与实验值较接近,结合能与实验值相差较远,广义梯度近似交换关联泛函得到的结果与实验值比其他两种泛函接近,所以选用GGA中的BPW91泛函[19]对Aln团簇的结构进行优化.
计算中考虑了不同的自旋多重度和各种可能的初始构型.为了验证得到的结构是真正的全局最小,在同等水平下计算了相应结构的振动频率,若发现存在虚频,则可沿最低虚频的振动方向调整原子坐标,直到找到一个真正的全局最小.
2 结果与讨论
在BPW91/LANL2DZ水平下研究了Aln(n=2~10)团簇的结构演化和磁性等,得到了Aln团簇的系列稳定结构、最低能量结构和亚稳态结构(图1).Aln团簇的结构参数见表2-3.

图1 优化得到的Aln(n=2~10)团簇的稳定结构
2.1 几何结构
Al2.Al2团簇为3重态,键长为0.285 8 nm,结合能为0.716 eV.Zhan等[20]在CCSD(T)/aug-cc-pVxZ水平下,计算得到Al2的基态结构为3重态.Sun等[21]运用B3LYP泛函计算得到Al2的基态结构也为3重态,键长为0.267 nm,结合能为0.653 eV.实验测得Al2团簇键长为0.270 nm,结合能为0.80 eV[18].所得Al2团簇的结合能与实验值较接近,说明所选方法可以准确描述团簇结构演化.
Al3.对Al3团簇,优化的结果表明在自旋多重度为2时具有D3h对称性的等边三角形能量最低如图1中3a所示,边长为0.261 nm,结合能为1.101 eV;Sun等[21]得到Al3团簇的基态结构也为等边三角形,边长为0.254 3 nm,结合能为1.058 eV.
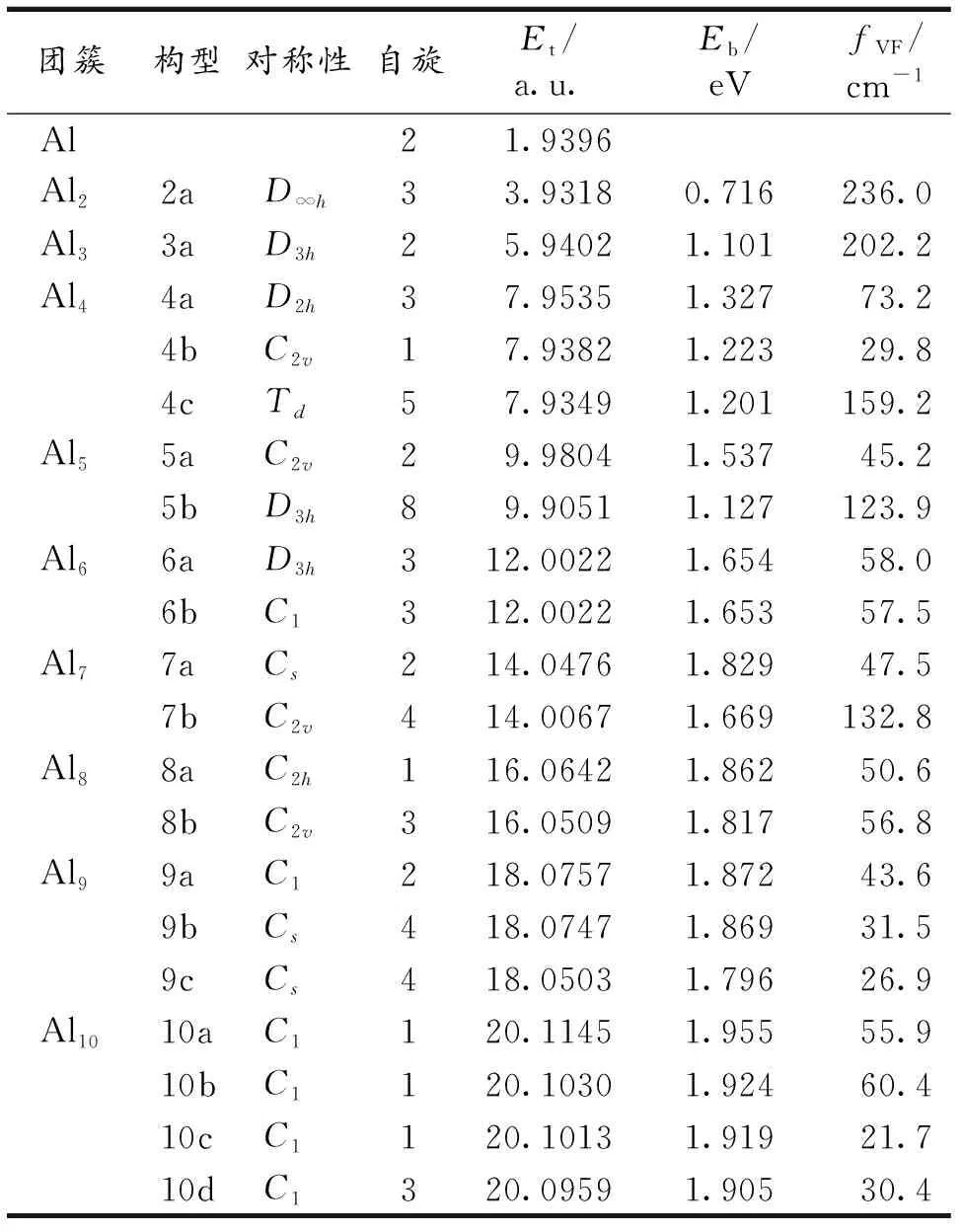
表2 BPW91/LANL2DZ水平下优化的Aln(n=1~10)
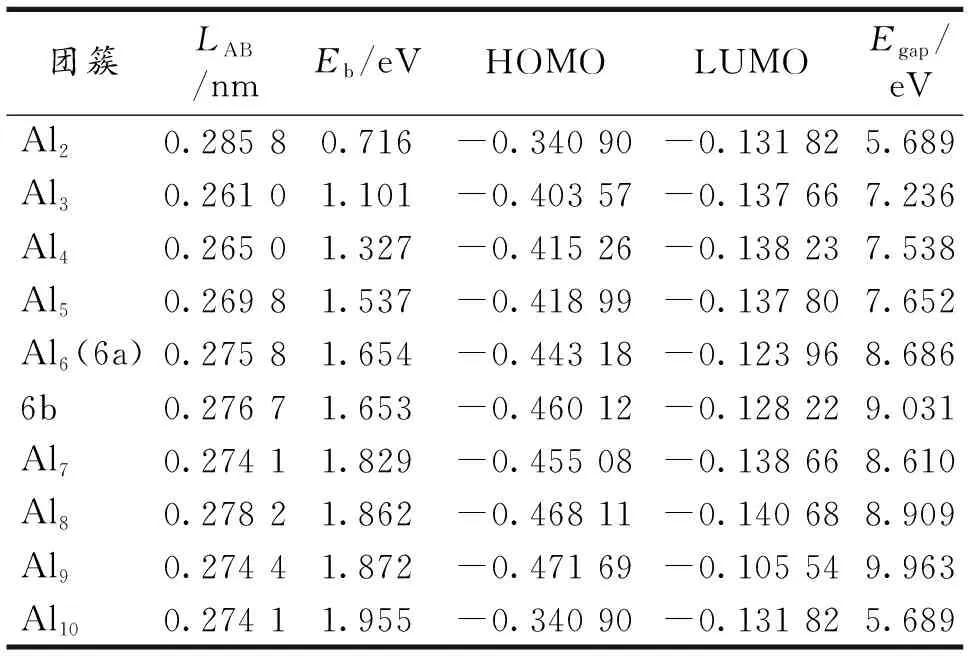
表3 Aln(n=1~10)团簇最稳定构型的平均键长LAB(nm)、 平均结合能Eb(eV)和能隙Egap(eV)
Al4.Al4团簇的最稳定结构为具有D2h对称性的菱形结构,如图1中4a所示,其自旋多重度为3,平均键长为0.265 nm,结合能为1.327 eV.第二稳定结构为具有Td对称性的四面体结构,如图1中4b所示,为一个自旋5重态,结合能为1.201 eV,能量比菱形高0.416 eV.具有C2v对称性的Y形结构是第三稳定结构,如图1中4c所示,为一个自旋单态,结合能为1.223 eV,能量比菱形结构高0.506 eV,比四面体结构高0.089 eV.Sun等[21]得到Al4团簇的基态结构也为菱形结构,边长为0.258 nm,结合能为1.229 eV.
Al5.对Al5团簇,具有C2v对称性的平面三角形两戴帽结构的自旋2重态为最稳定结构,如图1中5a所示,平均键长为0.269 8 nm,结合能为1.537 eV.具有D3h对称性的三角双锥结构的自旋8重态是一个亚稳态结构,如图1中5b所示,其结合能为1.127 eV,能量仅比5a高2.049 eV.Rao等[8]及Sun等[21]所得基态结构与5a相同.
Al6.Al6团簇得到了2个能量几乎简并的基态结构,分别为具有D3h对称性的三棱柱结构和具有C1对称性的畸变的八面体结构,分别如图1中6a,6b所示,三棱柱结构6a的平均键长为0.275 8 nm,结合能为 1.654 eV,八面体结构6b的平均键长为2.767 nm,结合能为1.653 eV,两个结构都为3重态.Rao等[8]及Sun等[21]得到Al6团簇的基态结构也为扭曲的八面体结构,D3h对称性的三棱柱结构6a为基态结构.
Al7.Al7团簇的最稳定构型为具有Cs对称性的扭曲的面心戴帽八面体结构,如图1中7a所示,此结构可看作是在八面体结构6b的基础上增加一个Al原子得到.其平均键长为0.2741 nm,结合能为 1.829 eV.具有C2v对称性的五角双锥结构7b是优化得到的第二低能异构体,它为1个自旋4重态,结合能为1.669 eV,能量比戴帽八面体结构7a高1.113 eV.Sun等[21]得到Al7团簇的基态结构也为扭曲的面心戴帽八面体结构,与笔者的结果一致.
Al8.优化得到Al8团簇的2个稳定结构都在八面体结构的基础上增加2个原子得到.最稳定结构为相对的2个面戴帽的具有C2h对称性的八面体结构,如图1中8a所示,为一个自旋单态,平均键长为2.782 nm,结合能为0.1862 eV.8b为第二稳定结构,结合能为0.1817 eV,为一个自旋三重态,能量比八面体结构8a高0.362 eV.Sun等[21]得到Al8团簇的基态结构与8b结构相似,与8a构型不同.
Al9.对Al9团簇,最稳定结构可看作是面心三戴帽八面体结构,如图1中9a所示.自旋多重度为2,平均键长为2.744 nm,结合能为0.1872 eV.9b和9c结构都具有Cs对称性,2个结构中两个Al原子为八面体面心的2个帽,另一个Al原子位置与四角面在同一个平面上.2个结构都为4重态.9b结构的结合能为0.1869 eV,能量比最稳定结构9a高0.027 eV.9c结构的结合能为1.796 eV,能量比最稳定结构9a高0.691 eV,比9b结构高0.664 eV.Sun等[21]得到Al9团簇的基态结构是在8a结构基础上增加一个原子得到,但与9a构型不同.
Al10.Al10团簇的最稳定结构为一个自旋单态,如图1中10a所示,平均键长为2.741 nm,结合能为0.195 5 eV.得到的另2个次稳定结构10b,10c都是自旋单态,结合能分别为0.1924 eV和 0.191 9 eV,这2个结构的能量分别比10 a高0.279 eV和0.359 eV.第四个次稳定结构10d为一个自旋三重态,结合能为0.190 5 eV,能量比10a高0.506 eV.Sun等[21]所得Al10团簇的基态结构与10a构型不同.
从上述对结构的分析中可以看出,Aln团簇的最稳定结构在原子数为n=6时经历了从平面结构到立体结构的转变,对小的Aln(n=2~5)团簇,最稳定结构都是在三角形的基础上增加一个原子生成的,且不能显著改变原有团簇的基本构型.随着原子数的增加基态构型都是在八面体的基础上增加原子得到的.
2.2 稳定性分析
为了分析团簇的稳定性随团簇尺寸的演化关系,我们分析了Aln团簇的平均结合能Eb、能量二阶差分D2(En)和分裂能Δ(En)随团簇尺寸的演化规律,其定义为Eb=(nE1-En)/n, D2(En)=En+1+En-1-2En, Δ(En)=(En-1+E1-En),其中n为Al团簇中所含Al原子的个数,En为Aln团簇的总能量,E1为单个Al原子的能量;并将平均结合能,能量二阶差分和分裂能随团簇尺寸的演化规律绘制在图2-4中.
从图2可以看到平均结合能Eb随团簇尺寸的增加表现出单调递增的趋势,且增幅随团簇尺寸的增加在逐渐减小.所作的平均结合能Eb随团簇尺寸的演化曲线与文献[21]所得Aln团簇的平均结合能随团簇尺寸的变化趋势相同.
在团簇物理中,能量二阶差分可以确切反映团簇稳定性.在图3中给出了能量二阶差分D2(En)随团簇尺寸的演化曲线.从图3可以看到Aln团簇的D2(En)在n=2~7时表现出明显的奇偶振荡行为,当n=3,5,7时能量二阶差分出现局域峰值,表明这些团簇较临近团簇稳定.n=7时能量二阶差分的峰值最高,说明所对应的团簇相对于其它团簇具有较高的稳定性.
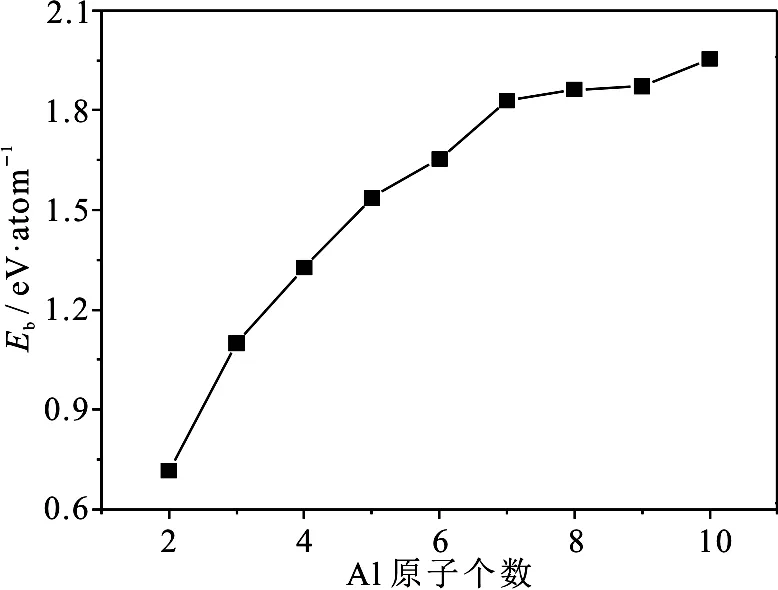
图2 Aln团簇的平均结合能Eb随团簇尺寸的演化
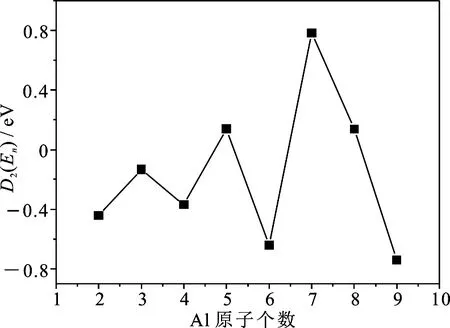
图3 Aln团簇的能量二阶差分D2(En) 随团簇尺寸的演化
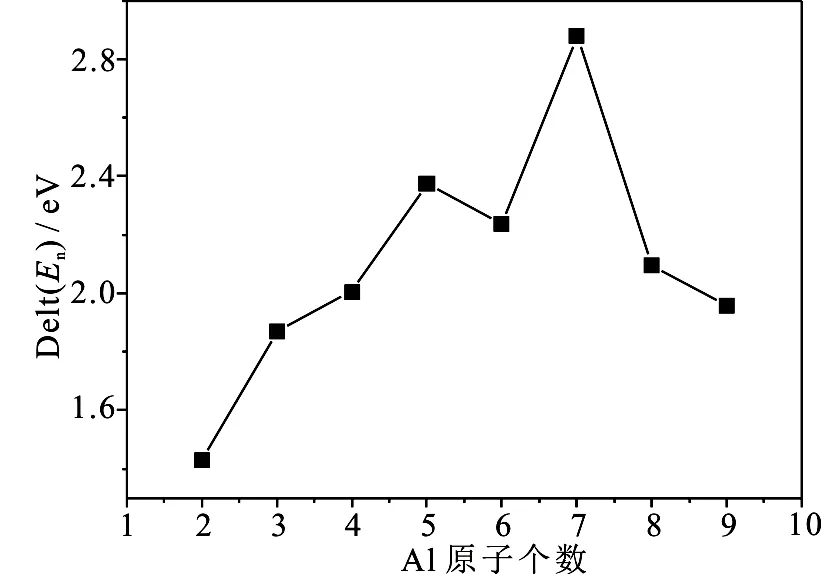
图4 Aln团簇的分裂能Δ(En)随团簇尺寸的演化
为了进一步分析团簇的稳定性与团簇尺寸的关系,分析了团簇的分裂能随团簇尺寸的变化.图4给出了团簇的分裂能Δ(En)随团簇尺寸的演化曲线,从图4可以看到,Aln团簇的分裂能随团簇尺寸的增加没有表现出明显的奇偶振荡行为,但在n=7时,分裂能的峰值最高,说明Al7团簇相对其它团簇具有较高的稳定性.
HOMO-LUMO能隙的大小反映了电子从占据轨道向未占据轨道发生跃迁的能力,在一定程度上代表团簇分子参与化学反应的能力,能隙越大,化学活性越弱.对小团簇而言,HOMO-LUMO能隙是衡量团簇相对稳定性的一个重要参数,能隙越大,相对应的团簇的稳定性越好.图5给出了团簇的HOMO-LUMO能隙随团簇尺寸的演化曲线.从图5可以看出HOMO-LUMO能隙在n=7时出现局域极大值,表明Al7团簇较相邻尺寸的团簇的稳定性要高.

图5 Aln团簇的能隙Egap(eV)随团簇尺寸的演化
2.3 磁性分析
图6 Aln团簇的平均每原子磁矩随团簇尺寸的演化
3 结束语
采用密度泛函理论中的广义梯度近似泛函(GGA)作为交换关联泛函详细研究了Aln(n=2~10)团簇的几何结构和磁性等.得到的如下结论:Aln(n=2~5)团簇的稳定构型都是在三角的基础上增加原子得到的,随着原子数的增加稳定构型都是在八面体和十面体的基础上增加原子得到的.得到的基态结构在n=6时从平面结构转变为三维结构,随着原子数的增加,逐渐形成笼状结构.团簇能量的二阶差分、分裂能、HOMO-LUMO能隙随团簇尺寸的演化都没有表现出明显的奇偶振荡行为,但在n=7时均有较大的值,说明相对应的团簇具有较高的稳定性.对团簇磁性的研究表明偶数个原子的团簇的平均原子磁矩为2μB,奇数个原子的团簇的平均原子磁矩为1μB,n=8,10时团簇的平均原子磁矩为0,与Rao等[8]结果符合.
猜你喜欢
大学物理(2022年9期)2022-09-28
教学考试(高考化学)(2022年4期)2022-08-30
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
陶瓷学报(2021年3期)2021-07-22
数学物理学报(2021年3期)2021-07-19
物理通报(2020年7期)2020-07-01
新世纪智能(数学备考)(2019年9期)2019-10-16
原子与分子物理学报(2015年3期)2015-11-24
