
利用CRISPR/Cas9基因编辑技术建立FcγR基因大片段敲除小鼠模型
2019-10-31 03:59吴曦霍桂桃刘甦苏谷文达曹愿柳全明吕建军范昌发
中国实验动物学报 2019年5期
吴曦,霍桂桃,刘甦苏,谷文达,曹愿,柳全明,吕建军,范昌发*
(1. 中国食品药品检定研究院实验动物资源研究所,国家啮齿类实验动物种子中心,北京 102629; 2. 中国食品药品检定研究院国家药物安全评价监测中心,北京 100176)
遗传修饰动物模型是基础和应用科学研究中不可或缺的工具,对其构建方法的改进从未停止。从最为经典的ES细胞介导的同源性重组,到各种DNA内切酶系统,如TALEN、ZFN和CRISPR/Cas9等技术,均先后被用于构建遗传修饰动物模型[1-2]。由于CRISPR/Cas9基因编辑技术具有特异性高、构建方便快捷、成本低廉、实验周期短等优势,正逐步成为基因组遗传修饰的主流技术。利用CRISPR/Cas9基因编辑技术实施基因敲除主要是通过一段20 nt的sgRNA引导Cas9蛋白结合于基因外显子区,Cas9对DNA切割后形成的双链断裂,通过非同源性末端连接造成随机碱基插入/缺失,导致基因发生移码突变[3]。也有部分研究使用两个sgRNA在基因组上同时进行切割,达到破坏基因表达的目的。该种方法的切割长度通常在数百至1000碱基左右[4]。然而在实际研究中,对基因簇、长链非编码RNA(long non-coding RNA, lncRNA)、基因调控序列的敲除往往涉及数千甚至数万碱基的敲除[5-6]。但CRISPR/Cas9基因编辑技术在大片段敲除方面的应用报道尚不多见,有待继续探索。
FcγR基因家族在造血系统中广泛表达,在调控体液免疫耐受、抗体依赖性的细胞免疫反应等多个方面具有重要作用[4],其表达异常与多种自身免疫性疾病相关。然而,由于物种间FcγR基因家族的结构差异及表达模式差异,从小鼠FcγR基因研究中得到的结果并不能完全反映其在人体中的功能[7]。因此,亟待建立导入人FcγR基因簇的人源化小鼠。另一方面,为了消除小鼠FcγR基因表达的潜在干扰,需要失活小鼠FcγR基因簇。小鼠中FcγR家族共包括四个基因:FcγR1、FcγR2b、FcγR3和FcγR4。其中,FcγR1位于3号染色体,大小为11 kb;FcγR2b、FcγR3和FcγR4形成基因簇,位于1号染色体106 kb范围内[8]。本文使用CRISPR/Cas9基因编辑技术对此基因簇进行敲除,通过在基因簇两侧设计sgRNA,通过活性验证后体外转录,与Cas9 mRNA共同注射受精卵,成功敲除了两个sgRNA之间90 kb片段,达到了对FcγR2b、FcγR3和FcγR4基因簇完全敲除的目的。通过测序验证,未发现脱靶现象,该敲除小鼠能稳定传代,可用于后续人源化小鼠模型的构建。
1 材料与方法
1.1 材料
1.1.1 实验动物
4 ~ 6周龄C57BL/6小鼠,由中检院实验动物资源所繁育并提供【SCXK(京)2017-0005】,饲养于本单位实验动物资源研究所屏障环境中【SYXK(京)2016-004】。饲养期间给予小鼠标准饲料及洁净饮用水(饲料购自北京科奥公司,饮用水由本单位经高压灭菌制备)。饲养环境:温度控制于18~26℃之间,湿度恒定,自动光控(昼夜各半交替)。所有操作均符合本单位实验动物伦理学要求(伦理审批号:#2017-B-004)。
1.1.2 试剂及仪器
(1)试剂。本研究所用CRISPR/Cas9活性检测试剂盒购自百奥赛图公司,实验所用引物均由诺赛基因公司合成。另外本试验所用其他各类试剂盒(如Cas9转录试剂盒、mMESSAGE、mMACHINE SP6、Transcription Kit等)和试剂(如高保真聚合酶、连接酶、DNA marker等)分别购自Life Technologies公司和宝生物工程大连有限公司(TaKaRa)[9]。
(2)主要仪器。电子天平(Sartorius,BSA220ZS型),离心机(美国Thermo Fisher公司),PCR仪(ABI公司),96孔板发光检测仪(Promega公司),NanoDrop 2000/2000C超微量紫外可见分光光度计(美国Thermo Fisher公司),显微注射仪(Eppendorf公司)等。
1.2 方法
1.2.1 候选sgRNA获取及切割位点载体构建
通过在线网站http://crispr.mit.edu设计获得候选sgRNA序列(见表1)。将合成的sgRNA引物配制为100 μmol/L,然后取上下游引物各10 μL混匀并放入沸水浴中5 min,水浴中自然降至室温退火。取3 μL退火后的引物与pCS载体(由CRISPR/Cas9活性检测试剂盒提供)进行连接,室温孵育2 h。
以野生型小鼠基因组为模板,利用Pybobest高保真DNA聚合酶及引物FcγR2b target f、FcγR2b target r、FcγR3 target f和FcγR3 target r进行切割位点扩增。扩增程序:95℃ 5 min、95℃ 30 s、55℃ 30 s、72℃ 30 s,共30 个循环。经PCR扩增后进行琼脂糖凝胶电泳,回收产物后取3 μL与pUCA载体于4℃连接过夜。
将连接好的产物转化至50 μL感受态细胞中,置于冰上孵育20 min,42℃热激细胞60 s,随后将转化产物加入SOC培养基中,37℃培养45 min后涂于平板。次日挑选克隆并接种至LB培养基,孵育8 h后抽提质粒后测序。

表1 候选sgRNA序列Table 1 Sequences of the candidate sgRNAs
1.2.2 活性检测
首先铺种293T细胞于96孔培养板中,然后将90 ng/孔sgRNA质粒(待检测sgRNA和试剂盒中提供的阳性对照sgRNA)与10 ng/孔切割位点质粒混合,加入1.2 μL PEI 转染试剂后混匀,并于室温放置15 min后加入96孔培养板中,5% CO2、37℃孵育24 h,随后利用Promega 96孔板发光检测仪进行荧光强度检测。
1.2.3 sgRNA的体外转录
体外sgRNA的转录方法见文献[9],即以sgRNA质粒为模板,利用引物T7-sgRNA f, T7-sgRNA r(见表2)进行PCR扩增。扩增程序:95℃ 5 min、95℃ 30 s、55℃ 30 s、72℃ 15 s,共30 个循环。然后将2倍体积的无水乙醇与PCR产物混合,以12 000 r /min离心10 min 后吸弃上清液,再加入500 μL 70%乙醇洗涤2次,随后将沉淀溶解于 30 μL RNase free的水中。利用MEGAshortscript T7 Transcription Kit在37℃环境下体外转录sgRNA 4 h,之后加入DNase并在37℃下消化20 min。然后利用MEGAclear Transcription Clean-Up Kit对消化产物进行纯化。对纯化后的产物进行浓度测定并分装,-80℃保存备用。

表2 PCR引物序列Table 2 Primer sequences for PCR
1.2.4 Cas9 mRNA的体外转录
Cas9 mRNA的体外转录与sgRNA的体外转录相似,即以px330质粒为模板,利用引物SP6-Cas9 f和SP6-Cas9 r(见表2)进行PCR扩增。扩增程序:95℃ 5 min、95℃ 30 s、55℃ 30 s、72℃ 2 min,共30个循环。将2倍体积无水乙醇与扩增产物混合,并以12 000 r/min离心10 min。弃上清后加入500 μL 70%乙醇洗涤2次,随后将沉淀溶解于30 μL RNase free的水中。利用mMESSAGE、mMACHINE SP6 Transcription Kit在37℃环境下体外转录Cas9 mRNA 1.5 h,之后加入Dnase并在37℃下消化20 min。然后利用Poly(A) Tailing Kit在37℃下进行加尾45 min。最后用RNeasy Mini Kit进行纯化。对纯化后的产物进行浓度测定并分装,-80℃保存备用。
1.2.5 胞质注射及胚胎移植
(1)超数排卵。选用4 ~ 6周龄C57BL/6雌鼠进行超数排卵处理,即在显微注射前 3 d 16:00时注射PMSG,前1 d 16:00 时注射hCG。随后将C57BL/6雌鼠与同品系雄鼠合笼,次日早晨检查阴道栓。将雌鼠于显微注射当日解剖,将受精卵从输卵管膨大部中取出,并将其以透明质酸酶消化为裸卵,随后将消化后的受精卵置于M2培养液中,以37℃、5% CO2培养备用。
(2)准备假孕鼠。挑选足够数量处于发情期的KM雌鼠,并将其于显微注射前1 d下午与结扎的同品系雄鼠以1∶1合笼。将雌鼠于显微注射当日早晨取出并查看有无阴道栓,把见栓的雌鼠作为0.5 d假孕雌鼠备用。
(3)显微注射及胚胎移植。移植前将sgRNA和Cas9 mRNA置于冰上解冻,将浓度为50 ng/μL的sgRNA 和100 ng/μL Cas9 mRNA混合,并以12 000 r/min离心20 min,吸取上清。操作时先用口吸管吸取液体,然后将液体从注射针后端注入尖端进行显微注射。将显微注射成功的囊胚放入胚胎培养液中并在37℃、5% CO2环境下培养,恢复30 ~ 60 min后,尽快将胚胎移植到0.5 d假孕雌鼠的子宫内。

注:FcγR2b、FcγR3和FcγR4基因位于小鼠1号染色体长臂,此基因簇长度为106 kb, 在基因簇两侧的FcγR3和FcγR2b基因内设计sgRNA切割位点。图中粗竖线代表基因外显子。图1 FcγR基因簇结构及sgRNA切割位点Notes. FcγR2b, FcγR3 and FcγR4 are located in the long arm of mouse chromosome 1. The length of this gene cluster is 106 kb. SgRNA cleavage sites were designed in FcγR3 and FcγR2b genes flanking the gene cluster. The thick vertical lines in the figure represent the exons of the gene.Figure 1 Structure of FcγR gene cluster and cleavage sites of sgRNA
1.2.6 靶位点扩增及序列测定
逐只提取首建鼠尾部基因组DNA并通过PCR方法检测基因型。引物设计针对转基因的编码序列,上游引物为FcγR2 target f,下游引物为FcγR3 target r(见表2)。扩增程序:95℃ 5 min、95℃ 30 s、55℃ 30 s、72℃ 30 s,共35个循环。将扩增产物送至诺赛基因公司进行序列测定,测序引物为FcγR2b target f。
1.2.7 脱靶位点扩增及测序
对首建鼠实施剪尾并提取基因组DNA,取100 ng基因组DNA,对所预测的8个脱靶位点进行PCR(见表3)。扩增程序:95℃ 5 min、95℃ 30 s、55℃ 30 s、72℃ 30 s,共35个循环。将PCR产物进行琼脂糖凝胶电泳后观察并切胶回收,然后将回收产物连接至pMD-18T载体并进行转化。挑4~5个克隆孵育后提取质粒,随后送诺赛基因公司测序,测序引物为M13F。

表3 脱靶位点PCR扩增引物Table 3 Primer sequences for PCR amplification of off-target site products
2 结果
2.1 切割位点设计
FcγR2b、FcγR3和FcγR4基因位于小鼠1号染色体长臂,三者所形成的基因簇分布于106 kb范围内(见图1)。因此在设计切割位点时,首先在FcγR2b外显子6和FcγR3外显子3的位点处各选取一个长度为250 bp的序列,随后利用美国麻省理工大学张峰教授实验室提供的预测软件(http://crispr.mit.edu/)设计sgRNA。通过软件预测总共得到40个可选用的sgRNA,然后在两个切割位点再分别选取评分最高,即脱靶效应概率最低的5个sgRNA进行活性检测。
2.2 体外活性检测
利用CRISPR/Cas9活性检测试剂盒对上述预测的两个切割位点的10个脱靶效应概率最低的sgRNA进行检测,即对每个sgRNA的体外活性进行 3 次独立试验。将检测荧光值同阳性对照荧光值比较后发现,多数sgRNA体外切割活性均高于阳性对照荧光值的60%,提示其在体内也有较好切割活性(见图2)。
2.3 制作大片段敲除小鼠
我们对FcγR2b4# sgRNA和FcγR3 3# sgRNA进行体外转录,并将sgRNA mRNA和Cas9 mRNA通过显微注射至受精卵并进行胚胎移植(见图3)。本研究共注射受精卵585个,注射后存活并移植胚胎300个,得到出生小鼠56只。

注:使用CRISPR/Cas9活性检测试剂盒进行sgRNA活性检测,sgRNA活性表示为转染候选sgRNA细胞荧光值与转染阳性对照sgRNA细胞荧光值之比。对于FcγR2b和FcγR3两个基因位点而言,位于左侧的sgRNA活性最高,首选用于注射。图2 sgRNA的体外相对活性Notes. The activity of sgRNA was detected by CRISPR/Cas9 activity detection kit. The activity of sgRNA was indicated by the ratio of fluorescence value of sgRNA transfected candidate cells to that of sgRNA transfected positive control cells. As far as the two loci of FcγR2b and FcγR3 were concerned, the sgRNA on the left had the highest activity and was selected for injection.Figure 2 Relative activities of sgRNAs in vitro
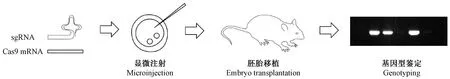
注:FcγR2b、FcγR3和FcγR4敲除小鼠构建流程:将sgRNA及Cas9 mRNA注射受精卵并进行胚胎移植,在F0代小鼠出生后4-7 d进行剪尾并提取基因组DNA,在切割位点两侧设计引物进行基因型鉴定。图3 FcγR敲除小鼠构建流程Notes. Construction process of FcγR2b, FcγR3 and FcγR4 knockout mice: sgRNA and Cas9 mRNA were injected into fertilized eggs and performed embryo transfer. The tails of F0 generation mice were cut and genomic DNA were extracted at 4-7 days after birth. Primers were designed on both sides of the cleavage site for genotyping.Figure 3 Construction process of the FcγR gene knockout mice
假如Cas9蛋白对sgRNA靶位点进行了切割,那么切割位点则会发生非同源性重组以进行修复,并引起部分碱基插入或缺失,同时也存在两个切割位点间片段发生完全缺失的小概率情况。为了检测基因组中是否发生大片段缺失,我们使用两个切割位点左侧及右侧引物进行扩增。在56只小鼠中,有1只小鼠经PCR检测为阳性。进而,我们对PCR条带进行测序(见图4),确认导致了大小为89 711 bp的碱基缺失,造成Fcgr2b5’端,Fcgr3 3’端及Fcgr4整个基因簇缺失。

图4 FcγR敲除阳性小鼠测序结果Figure 4 Sequencing results of the positive FcγR knockout mice
2.4 脱靶位点检测
由于sgRNA存在一定脱靶效应,即发生错配并在基因组中形成非特异性结合。因此我们通过软件对两个用于注射的sgRNA的脱靶位点进行了预测(见表4),并对首建鼠中是否存在脱靶效应引起的突变进行了检测。分别选取4个脱靶概率最大的位点进行PCR扩增后构建T载体,挑取4~5个克隆孵育后抽提质粒进行测序。测序结果显示所有克隆均在脱靶切割位点测序正确(见图5),表明首建鼠中无脱靶效应造成的突变。

表4 脱靶位点序列及PCR检测结果Table 4 Off-target site sequences and PCR results

注:对8个预测脱靶位点分别进行PCR后连接T载体,挑取4-5个克隆进行测序并与参考序列进行比对,结果显示在sgRNA切割位点周边无小片段插入缺失。图5 验证脱靶效应的代表性测序图谱Notes. Eight predicted off-target sites were separately ligated to the T vector after PCR, and then 4-5 clones were picked for sequencing and aligned with the reference sequence. The results showed that there was no small fragment insertion of deletion around the sgRNA cleavage site.Figure 5 Representative sequencing maps to verify the off-target effects
2.5 敲除位点的稳定遗传
为了确认敲除位点能够稳定遗传,我们将首建鼠与C57BL/6背景雌鼠进行交配,共得到F1子代18只,其中阳性鼠7只,通过对其进行卡方检验P=0.44,表明其阳性率符合预期,提示这一敲除位点能够被稳定遗传。实际传代结果也显示,该小鼠表型与野生型小鼠无异,能正常建系传代,目前已经获得纯合子。
3 讨论
FcγR基因家族表达于不同种类的免疫细胞中,通过激活或抑制性受体平衡免疫反应。近年来,FcγR基因介导的抗体依赖细胞毒性作用(antibody-dependent cellular cytotoxicity,ADCC)广泛用于抗肿瘤治疗[10-11]。然而,小鼠与人类FcγR基因在表达模式及基因结构方面均存在较大差异,因而近交系小鼠难以模拟人类中的免疫反应。构建FcγR人源化小鼠将为相关研究提供很大帮助,而且构建FcγR基因敲除背景的小鼠是完成人源化的第一步[12]。小鼠中的FcγR2b、FcγR3和FcγR4形成基因簇,位于1号染色体106 kb范围内。由于基因间距离较近,很难通过传统方式分别构建3个基因敲除小鼠,再将其进行交配的方式得到3个基因敲除小鼠。因此,本研究采用CRISPR/Cas9基因编辑技术完全敲除了基因簇所在区域。
自2013年1月以来,多篇有关CRISPR/Cas9基因编辑技术的文章发表于Science、Nature、Cell等杂志,从而也证实其在哺乳动物细胞中具有DNA切割活性后[13],该技术被广泛应用于多种类型的基因组修饰,如敲除、敲入、转录激活、转录抑制等[14]。特别在基因敲除模型的构建方面,相较于传统ES打靶技术,CRISPR/Cas9基因编辑技术的优势更为明显。使用CRISPR/Cas9基因编辑技术进行基因敲除主要基于如下过程:即在预先设计好的sgRNA指引下,Cas9蛋白对靶基因进行序列特异性地切割后造成双链断裂,随后被切割位点发生非同源末端连接(non-homologous end joining,NHEJ),此时通常会造成切割位点产生小片段插入或缺失,从而造成基因的移码突变[15-16]。由于以CRISPR/Cas9基因编辑技术进行基因敲除的方法不需要进行同源性重组,因此具有敲除效率高的优势,并且使同时敲除多个基因成为可能。然而,由于这一方法通常只造成小片段插入或缺失,一方面难以使用常规PCR方法进行基因型鉴定,因此为大规模生产繁育造成困难。另一方面存在基因仍可正常转录的可能,不利于检测基因是否完全失活。在本研究中,为了解决上述两方面的问题,我们预先设计了两个sgRNA来引导Cas9蛋白在靶基因特异性切割位点两侧同时进行切割,引起切割位点之间的片段发生整体删除,为后续基因型鉴定提供了便利。
在染色体上制造大片段缺失具有广泛的应用价值,如模拟人类疾病中的大片段染色体异常,对基因簇如Hox基因家族、Mage基因家族等进行敲除等[17-18]。目前已知对小鼠基因组进行敲除的最大片段长约1.6 MB,但此研究仍使用ES打靶技术进行敲除,通过筛选阳性ES细胞进行囊胚注射得到[19]。在本研究中,通过使用由张峰实验室开发方法并在其网站http://crispr.mit.edu 对sgRNA进行设计,为了尽量避免脱靶效应,选择评分高的sgRNA作为候选sgRNA。根据先前的研究,即使切割位置较为接近的sgRNA切割效率也存在很大差异,因此我们首先进行体外活性检测,在两侧选取活性较高的sgRNA。在本研究中出生的56只小鼠中,有1只被确认为大片段敲除阳性,阳性率为1.78%。
CRISPR/Cas9的靶向识别序列长度为20碱基,再加上识别需要的前间区序列邻近基序(protospacer adjacent motif,PAM)位点。如果RNA与染色体靶位点的结合是严格以序列为依据,那么CRISPR/Cas9错误切割的可能性极低。然而根据报道,在存在一个或多个错配的情况下,sgRNAs仍然能够引导Cas9核酸酶作用于被整合到染色体上的GFP报告基因[20]。因此,在以CRISPR/Cas9系统构建的遗传修饰小鼠模型中进行脱靶位点的突变检测是必需的。本研究中根据网站http://crispr.mit.edu 对sgRNA脱靶位点的预测,就两个sgRNA概率最高的4个脱靶位点进行PCR测序,确认在首建鼠中无脱靶效应引起的突变。
综上所述,在本研究使用CRISPR/Cas9基因编辑技术构建得到FcγR2b、FcγR3和FcγR4基因簇敲除小鼠,敲除片段长度达90 kb。本研究一方面验证了CRISPR/Cas9基因编辑技术进行大片段敲除的可行性,为建立其他大片段敲除模型提供了快速便捷的方法;另一方面构建了FcγR2b、FcγR3和FcγR4基因簇敲除小鼠,经过近2年、传代5代以上,该小鼠模型能正常繁殖建系,并且遗传稳定,为下一步构建FcγR基因人源化小鼠奠定了基础。
致谢:作者感谢北京市第二十五中学吕云松在DNA提取和PCR实验中所提供的帮助。
猜你喜欢
军事文摘(2022年16期)2022-08-24
中国计量大学学报(2022年2期)2022-07-18
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
科技与创新(2020年12期)2020-11-28
中西医结合肝病杂志(2020年2期)2020-10-27
做人与处世(2020年10期)2020-06-29
肿瘤防治研究(2019年7期)2019-01-06
科技创新与品牌(2018年5期)2018-07-24
新农村(2018年35期)2018-04-02
