
铟掺杂钨位增强钨酸铋氧空位光催化效率*
2019-11-08 08:45王泽普付念于涵徐晶威何祺郑树凯丁帮福闫小兵
物理学报 2019年21期
王泽普 付念 于涵 徐晶威 何祺 郑树凯† 丁帮福‡ 闫小兵
1) (河北大学电子信息工程学院,保定 071002)
2) (河北大学物理科学与技术学院,保定 071002)
以硝酸铋、硝酸铟、钨酸铵、柠檬酸、聚乙二醇为原料和表面活性剂,通过溶胶−凝胶法合成纯的和铟掺杂正交晶系钨酸铋.X射线衍射表征得到所有样品都是纯相且无杂质相.光催化降解罗丹明B实验发现,铟掺杂样品降解能力高于纯相,其最佳掺杂含量为7%摩尔比.扫描电子显微镜和X射线光电子能谱表征发现,光催化性能提高主要是由于氧空位数目增多导致,而形貌发生蓬松改变起到促进作用.利用第一性原理计算,单一氧空位模型形成能小于Bi1In + VO和Bi2In + VO共掺杂模型形成能,而大于WIn + VO形成能.这个结果表示铟替代钨位促进氧空位产生.电子结构计算发现,氧空位在带隙和导带底附近引入新的局域态,促进光吸收而增强光催化性能.本文通过理论模拟和实验表征钨酸铋氧空位光催化性能调控归因于铟进入钨位而非铋位.
1 引 言
钨酸铋(Bi2WO6)是一种高效光催化剂,属于层状钙钛矿奥利威利斯型结构的一种[1].通过漫反射光谱测试,Bi2WO6禁带宽度大约在2.7 eV[2].由于合适的带隙值,钨酸铋可以进行紫外−可见光降解污染物作用.例如通过水热反应合成的三维分层钼掺杂钨酸铋,在氙灯照射下,5%掺杂样品降解罗丹B效率高达97%[3].因此最近几十年来,研究者通过各种方法合成不同微观形貌以及掺杂和复合策略改进钨酸铋光催化降解效率[4,5].
除了上述改性之外,钨酸铋中的氧空位在催化过程中也起到了重要作用[6].例如卢青等[7]采用乙二醇作为还原剂,利用溶剂热法制备含氧空位钨酸铋纳米材料.通过降解气相苯以及扫描电子显微镜(SEM),Mott−Schottky等表征,含氧空位钨酸铋催化性能的增强主要来源于氧空位缩小其带隙宽度,提高光吸收强度.许雪棠等[8]利用水热法制备出含氧空位钨酸铋,光催化降解10 mg/L罗丹明B的效率高达98.84%.此外氧空位能够拓宽钨酸铋光响应吸收范围,例如Lü等[9]采用水热法合成钨酸铋粉末,然后在氦气和氢气 + 氩气环境下进行热处理制备出含氧空位钨酸铋(Bi2WO6-x),经后续热处理的样品,可见光响应从450 nm扩展到600 nm.Bi2WO6-x样品光降解2−4−二氯苯酚效率大约是原始Bi2WO6的2.1倍.综上可见,钨酸铋中氧空位起到移动带边以及伴随大量氢氧根基团从而增强光响应和增加氢氧根自由基活性位置作用[10].尽管氧空位在光催化中起到至关重要作用,但上述方法不能准确调控氧空位浓度[11,12].
元素掺杂是一种简单控制氧空位浓度方法.例如引入锆元素,改变锆元素含量,催化性能逐渐提高,最佳含量为3%,其主要原因是锆掺杂促进了氧空位的产生[13].此外水热法合成铜离子修饰的钨酸铋降解性能要高于纯的钨酸铋,其原因是由于铜离子通过多电子转移机制减少氧离子而产生氧空位[14].上述方法能够调控氧空位浓度,并且通过实验表征能给出一定的机理解释,但缺少理论支持.本文通过铟掺杂钨酸铋实现氧空位的简单调控,并通过形成能计算给出铟掺杂增强氧空位的理论解释.因此,本文实验与理论相结合的方法可以推广到其他光催化剂的机理研究.
2 实验过程和理论计算
将1.21 g五水硝酸铋和1.58 g柠檬酸混合溶于一定浓度稀硝酸中,磁力搅拌至透明溶液,标记为A溶液.将0.355 g钨酸铵放入去离子水中,加热并磁力搅拌溶解至透明,标记为B溶液.待溶液B冷却至室温后,利用滴定管,将其缓慢滴入到溶液A中,并在滴定过程中进行剧烈磁力搅拌,最终混合溶液标记为溶液C.在溶液C中加入少量聚乙二醇作为表面活性剂,并利用稀硝酸和氨水将pH值调节为1.0,得到溶液D,最后将其转移到80 ℃水浴锅中,水浴一定时间后,溶液中将产生乳白色絮状络合物,将其置入120 ℃烘箱中干燥,取出后再于450 ℃下煅烧3 h,自然冷却到室温并进行研磨以备后续测试.不同浓度铟掺杂钨酸铋制备过程同上,只是在溶液A中按化学计量比加入硝酸铟.整个制备过程示意图如图1所示.
利用丹东通达科技有限公司生产的TD−3500型X射线衍射仪(XRD)对样品进行晶体结构表征;利用Zeiss supra 55型SEM对样品的微观形貌进行表征;采用赛默飞Escalab 250 Xi光电子能谱仪对样品进行X射线光电子能谱(XPS)测试.光催化效果测试过程为: 将50 mg样品放入烧杯中,再取50 mL浓度为5 mg/L的罗丹明B溶液放入烧杯中,在暗处磁力搅拌30 min达到吸附脱附平衡.然后用250 W汞灯光源进行光照,每隔15 min取样离心后采用北京普析通用仪器有限责任公司生产的TU−1901型双光束紫外可见分光光度计测定样品对罗丹明B吸光度C,通过(C-C0)/C得到降解效率曲线.
为了探索铟掺杂对钨酸铋催化性能的作用,采用第一性原理计算软件进行形成能和电子结构计算[15].所有计算采用截断能为400 eV,能量和原子受力收敛标准为1.0 × 10-4eV和-0.03 eV/atom.以纯单胞计算为例具体说明计算过程,首先建立钨酸铋单晶胞,然后进行全弛豫和自洽计算得到电荷密度,最后基于电荷密度计算能带和态密度.由于纯密度泛函理论计算得到的带隙值小于实验值,采用18%杂化密度泛函进一步计算修正带隙值.单胞中扣去一个氧原子形成单一氧空位模型.单胞中扣去一个氧原子并用铟替代钨或铋位形成掺杂 +氧空位模型.

图1 溶胶−凝胶法制备钨酸铋粉体实验流程图Fig.1.Schematic diagram of sol−gel method for bismuth tungstate powder preparation.
3 结果与讨论
图2(a)为纯相和铟掺杂钨酸铋XRD图谱,可以看出,纯相XRD与ISCD 67647 模拟衍射模式相吻合,表示正交单相钨酸铋合成.铟离子引入并没有改变衍射峰的位置和数目,且无其他杂质峰出现,从侧面说明铟离子进入钨酸铋晶格中.根据离子半径大小[16],铟离子最可能进入铋位,然而形成能比较发现,铟进入钨位的形成能小于进入铋位的形成能,说明铟进入钨位才更有利于氧空位的生成.而根据XPS测试,掺杂后材料中的氧空位确实增多,说明在本实验中,铟更有可能进入钨位.此外图中标出所有衍射峰对应的晶面,其中最强峰对应晶面为(13).图2(b)为优化后的纯钨酸铋单晶胞结构,可以看出,钨酸铋是由WO6和BiO3交替排列的层状结构.W有一种位置,Bi有两种位置,O有六种位置,根据Wcykoff坐标和空间群对称性,所有原子位置均为C1对称性.
图3显示了纯的和不同浓度铟掺杂钨酸铋微观形貌,可以看出,所有样品都是由纳米小颗粒聚集而成的大颗粒形貌.随着铟掺杂浓度提高,这种大颗粒形貌变得逐渐规则,如10 at%掺杂类似于球状.掺杂后样品形貌堆积变得更加蓬松.在金属掺杂时,有时所掺杂的原子或者其化合物会附着在材料表面,可能会影响到钨酸铋材料的结晶范围,这可能是导致掺杂后样品更加蓬松的原因.这种蓬松结构使得样品与液体接触面积增大,增强光催化降解活性位置.
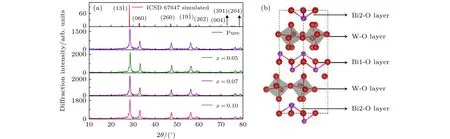
图2 ICSD模拟和不同浓度铟掺杂钨酸铋XRD图谱(a)和优化完正交钨酸铋结构模型(b)Fig.2.XRD patterns of the ICSD simulation and different concentration In−doped bismuth tungstate (a),as well as optimized orthogonal bismuth tungstate structure model (b).

图3 样品微观形貌结构图 (a)纯相,(b) 5 at%,(c) 7 at%,(d) 10 at% In−Bi2WO6Fig.3.Microscopic morphology of samples: (a) Pure phase,(b) 5 at%,(c) 7 at%,and (d) 10 at% In−Bi2WO6.

图4 不同铟掺杂钨酸铋的降解效率(a)和一级反应速率常数柱状图(b)Fig.4.Degradation efficiency of In−doped bismuth tungstate (a) and the first−order reaction rate constant (b).

图5 纯相(a)和7%铟掺杂(b) Bi2WO6的O 1 s XPS光谱以及Gaussian分峰Fig.5.O1 s XPS spectra and Gaussian peaks of pure phase (a) and 7% In−doped Bi2WO6 (b).
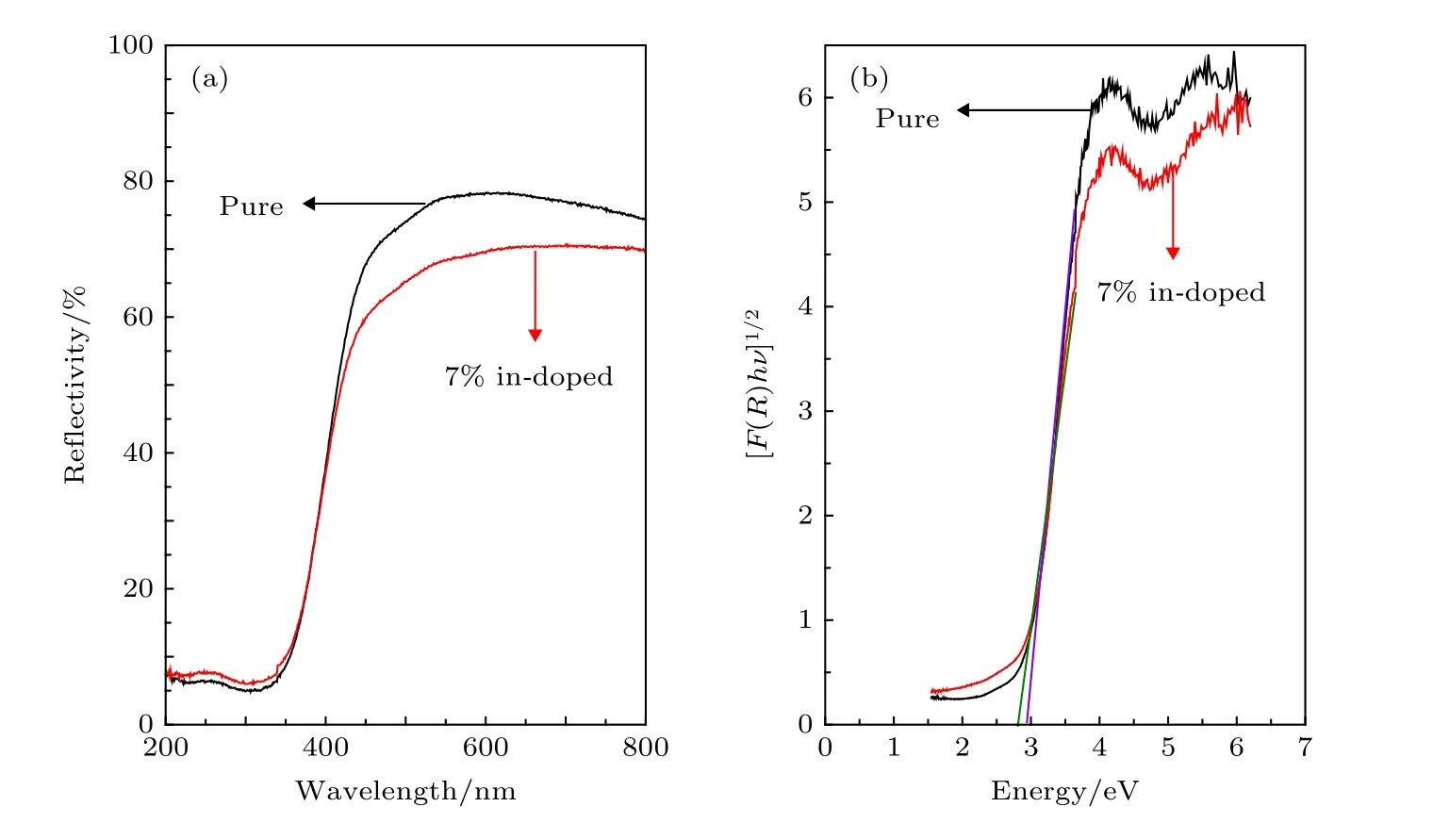
图6 纯的和7 at%铟掺杂样品的漫反射光谱(a)和K−M方程拟合的带隙值(b)Fig.6.Diffuse reflectance spectra of pure and 7 at% In−doped samples (a) and band gap values fitted by K−M equation (b).
图4(a)为纯相和不同浓度铟掺杂Bi2WO6材料的光催化降解罗丹明B曲线,可以看出,经过250 W汞灯90 min照射,纯相样品光催化效率约为75%,铟离子引入加快催化效率,例如7 at%铟掺杂样品约为90%.总之,铟掺杂样品催化效率都高于纯相.通过XRD和SEM分析可知,降解催化增强的主要原因并不是来源于形貌和结构变化.图4(b)给出了根据一级反应速率常数k=ln(C0/C)/t[17]得出的反应速率柱状图,可以看出,所有铟掺杂样品反应速率大于纯相样品.
为了探索铟掺杂增强催化性能的原因,首先对样品进行价态表征,结果如图5所示.纯相和铟掺杂样品O 1s XPS呈现非对称分布.通过高斯拟合得到3个峰: A,B,C.在纯相中,A,B,C峰位为530.053,530.693,531.584 eV.低能峰A和B归因于Bi−O和W−O配位[18],而高能峰C来源于缺陷氧.因此钨酸铋材料较高的催化性能主要来源于氧空位[19].铟掺杂导致高能峰C强度增大,表示铟替代晶格位置促进氧空位产生,因而导致掺杂样品催化效率高于纯相.

图7 (a)四种模型结构示意图及其(b)相应形成能变化趋势Fig.7.Schematic diagram of four model structures (a) and corresponding formation energy variation (b).

图8 纯相(a),(b)和单一氧空位(c),(d)模型电子结构 (a),(c) 能带图;(b),(d) 态密度图Fig.8.Pure phase (a),(b) and single oxygen vacancy (c),(d) model electronic structure: (a),(c) Energy band diagram;(b),(d) density of states (DOS).
图6(a)为纯相和7%铟掺杂钨酸铋紫外−可见光漫反射光谱,可以看出,纯样品在紫外区域有强吸收,而在可见光区域有20%吸收.通过铟掺杂使得样品在可见光区域吸收增大到30%.因而铟掺杂提高了样品的可见光响应,从而可能进一步提高光催化降解效率.通过Kubelka−Munk函数拟合可以得到样品带隙值,如图6(b)所示[20].掺杂前后,样品带隙值分别为2.79 eV和2.74 eV,带隙略有减小,进一步增强光催化降解过程中光吸收.
为了进一步从理论上确定铟掺杂提高氧空位浓度,分别建立了四种模型,即单一氧空位(VO)、铟代替铋位 +VO、铟取代钨位 +VO.弛豫后的四种模型如图7(a)所示.利用公式

计算四种模型形成能,如图7(b)所示,可以看出,单一VO形成能大约为4.25 eV,而BiIn+ VO形成能高于VO模型,表示铟替代铋位不利于氧空位产生.WIn+ VO模型形成能大约为2.25 eV,远低于VO模型,因此铟掺杂替代钨位促进氧空位浓度的增高,这个结果与XPS分析相符合.
图8所示为纯的和单一氧空位模型的电子结构,可以看出,由于杂化泛函计算,纯相钨酸铋带隙为2.76 eV,和实验值相符合.对比掺杂前后的能带图可以看到,铟掺杂后,能带中引入带隙态能级并导致导带边下移,使得材料光吸收能力增强.从态密度图可以看出,这些带隙态主要是钨5d组成.由于5d空态,因此铟掺杂导致空带数目增多,引起更多电子从价带跃迁到导带和带隙态,增强可见光吸收,从而提高光催化活性,这个结果与漫反射测试结果相一致.
4 结 论
本文通过溶胶−凝胶法制备纯相和不同浓度铟掺杂Bi2WO6纳米颗粒.XRD测试表明所有样品均为正交晶系的多晶结构,铟掺杂未引入其他杂质峰;SEM测试表明铟掺杂样品微观形貌由不规则形状堆积逐渐转化为类似球形堆积,且表面变得更加蓬松,暴露表面更大;漫反射光谱测试表明,铟掺杂导致带隙减小,进一步增强光吸收;XPS测试表明,铟掺杂致使材料氧空位数目增多,同时第一性原理计算进一步说明铟掺杂钨位使Bi2WO6材料中氧空位形成能减小,更有利于其生成.Bi2WO6光催化剂中氧空位增多,可以俘获更多电子,从而阻止催化过程中载流子复合,提高光催化效率.
猜你喜欢
物理学报(2022年17期)2022-09-14
成都信息工程大学学报(2019年3期)2019-09-25
传感器世界(2019年11期)2019-02-17
陶瓷学报(2019年5期)2019-01-12
科技创新与应用(2018年21期)2018-09-14
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04
陶瓷学报(2015年4期)2015-12-17
天津师范大学学报(自然科学版)(2014年2期)2014-08-06
读者欣赏(2014年6期)2014-07-03
