
Li离子电池负极材料石墨炔在B,N掺杂调控下的储Li性能优化*
2019-11-08 08:44蔡梦圆唐春梅3张秋月
物理学报 2019年21期
蔡梦圆 唐春梅3)† 张秋月
1) (河海大学浅水湖泊综合治理与资源开发教育部重点实验室,南京 210098)
2) (河海大学理学院,南京 210098)
3) (南京大学固体微结构物理国家重点实验室,南京 210093)
一种理想的Li离子电池负极材料需要具有较高的储Li容量和较低的体积膨胀比.本文应用密度泛函理论研究了二维多孔石墨炔在B,N原子掺杂调控后作为Li离子电池负极材料时的储Li性能.计算结果表明,B在石墨炔结构中的掺杂可以增强Li与石墨炔之间的吸附作用,储Li容量可以增加到2061.62 mAh/g,与未掺杂单层石墨炔相比增加了2.77倍.同时,B掺杂降低了Li在垂直于石墨炔平面方向上的扩散能垒,而面内扩散能垒提高了0.1 eV.N掺杂降低了Li与石墨炔之间的相互作用,但增加了Li的稳定位点,储Li容量增加到了1652.12 mAh/g,同时,Li在石墨炔上的扩散性能大大提高,在平面内扩散能垒降至0.37 eV,因此N掺杂石墨炔的充放电性能得到较好提升.因此,B,N掺杂可从不同方面提升石墨炔作为Li电池负极材料时的储Li性能.该研究可以为开发良好的储Li负极材料提供一个良好的研究思路,为实验工作者提供理论依据.
1 引 言
随着化石能源的消费和全球经济的不断增长,可再生清洁能源是未来的发展方向.先进的储能技术在电子设备[1]、电动车[2]、智能电网[3]中具有重要意义.可充电电池[4−6]、储氢介质[7]、超级电容器[8]等新型储能装置已经引起了人们的广泛关注.Li离子电池是常用的可充电离子电池之一,目前已广泛应用于许多设备中.一种理想的Li负极材料应该达到以下标准: 1) Li的平均吸附能(average adsorption energy,Ead)应小于3.0 eV[9],大于固体Li的实验内聚能(1.63 eV[10]);2)当Li电子电池的负极开路电压大于2 V时,负极将停止工作,如果开路电压为负值,容易在负极材料上发生析Li现象形成枝晶,影响充放电性能,因此负极的OCV值应该在0-2 V[11,12];3)负极材料在Li吸附过程中的体积膨胀比应尽可能小,避免Li离子电池负极充放电循环速率的快速衰减[13,14].
基于碳原子的可循环充电Li离子电池商业化之后,人们做了大量努力来提高碳负极材料的各项性能指标,如储Li容量和充放电速率.研究发现纯碳材料并不总是最适合的电极材料,但是,采用异性原子对纯碳材料进行掺杂会显著提高其作为负极材料时的储Li性能等[15].N原子和B原子是迄今为止研究最广泛的掺杂原子,因为N和B在元素周期表中与C相邻,使用它们掺杂之后可以明显提高碳材料的化学活性.目前已有许多关于N掺杂的石墨烯及其他二维材料储Li性能的研究[16−19].例如: Tian等[20]制备的N掺杂石墨烯片在100 mA/g的电流下可逆储Li容量能达到832.4 mAh/g,经过108次循环后可逆储Li容量仍保持750.7 mAh/g.Zhang等[21]在NH3气环境中进行热处理得到氮掺杂石墨二炔薄膜,在200 mA/g的电流下经过200次循环后可逆储Li容量为785 mAh/g,高于纯石墨二炔的可逆储Li容量(584 mAh/g).B掺杂的石墨烯和碳纳米管具有很好的氧化还原性能[22,23].Luo等[24]将B原子掺入石墨发现石墨中饱和Li密度增加33.3%.以上研究均表明N掺杂和B掺杂可以明显改善碳材料作为Li离子电池负极材料时的各项性能指标.
石墨炔是一种新型纯碳纳米材料,较早就由Baughman等[25]理论预测存在,但是近期才被成功实验合成[26].石墨炔比石墨烯具有更大的孔洞,因此被认为是更理想的Li离子电池负极材料.最近,研究表明,B,N掺杂的石墨炔结构特别稳定,会从半导体变成导体[27−29],因此具有较好的导电性能,理论上应该是更加理想的Li离子电池负极材料.但是,到目前为止,还没有人对B,N掺杂的石墨炔作为Li离子电池负极材料的储Li性能进行详细深入研究,因此,本文将深入详尽地探究B,N掺杂的石墨炔结构作为Li离子电池负极材料的可行性.
2 计算方法
该工作所有的计算采用基于密度泛函理论(density functional theory,DFT)[30]的DMol3软件[31,32]来完成.所有计算采用基于广义梯度近似[33]的Perdew−Burke−Ernzehof (PBE)交换关联函数[34]和全电子轨道DNP基组(double−numerical quality with inclusion of polarization functions)[31].此外考虑范德瓦耳斯相互作用,在计算中引入了基于Grimme的DFT−D2色散修正方法[35],建立了2 ×2 × 1的周期性晶胞模拟B,N掺杂的石墨炔二维材料.为了避免层间相互作用,在垂直于片层的方向上设置了20 Å的真空层.由文献[16−19]可知,20 Å的层间距足以避免二维材料的层间作用.布里渊区中K点设置为9 × 9 × 1,轨道截断半径设置为4.6 Å.在几何优化计算中,结构的弛豫收敛标准被设置为: 总能量的收敛标准为10-6Hartree,力的收敛标准为0.001 Hartree/Å及位移的收敛标准为0.001 Å.最低能量结构被确定为基态结构,并进一步通过声子谱计算确认无虚频来进一步进行验证.
3 结果与讨论
3.1 单个B,N原子掺杂的石墨炔结构
首先,选用石墨炔原胞进行B,N掺杂.为了尽可能地减少计算量,结构同时需要含有两个完整的孔洞,因此,选取2 × 2 × 1的石墨炔晶胞进行掺B,N研究,2 × 2 × 1的石墨炔晶胞的空间群为P1,晶格常数a=b=6.88 Å.石墨炔有两种不同类型的碳原子: 位于六元环(ring)上的sp2杂化C原子命名为C1,位于乙炔链(chain)上的sp杂化C原子命名为C2.因此B和N原子有两种替代掺杂位置,即环掺杂(替代C1原子)和链掺杂(替代C2原子),见图1.B和N掺杂优化结构a,b方向的晶格常数均相等,但是发生了改变,B环掺杂的石墨炔晶格常数a=b=6.98 Å,B链掺杂石墨炔晶格常数a=b=6.92 Å.N环掺杂石墨炔晶格常数a=b=6.86 Å,链掺杂石墨炔晶格常数a=b=6.90 Å,与之前报道一致[27].B和N的掺杂分别引入了两种新键,即B-C1和B-C2,N-C1和N-C2.表1列出了B,N掺杂之后石墨炔的晶格常数、键长、Mulliken电荷及B,N原子的结合能(binding energy,Eb).

图1 2 × 2 × 1的石墨炔晶胞中单个B,N的两种掺杂位点,分别为环掺杂和链掺杂Fig.1.Two doping sites of single B and N in the 2 × 2 × 1 supercell of graphyne.They are ring doping and chain dop−ing respectively.

表1 B,N掺杂的石墨炔的晶格常数、键长、Mulliken电荷及EbTable 1.Lattice constant,bond length,Mulliken charge and Eb of B,N doped graphyne.
由于B原子的半径比C原子大,B-C键要长于原来石墨炔中的C-C键长,由表1可知,环掺杂的B-C2键长和B-C1键长均为1.50 Å,小于B-C单键键长(1.52 Å),同时大于B-C双键键长(1.44 Å)[36].B替代掺杂石墨炔中C-C大π键形成的B-C1键长为1.54 Å,大于B-C单键键长(1.52 Å).而B链掺杂形成的B-C2键长为1.36 Å,小于B-C双键键长(1.44 Å)[36],但远大于B-C三键键长(1.23 Å)[37].由于N的原子半径较小,当N在石墨炔中替代掺杂时形成的N-C键长要短于石墨炔中的C-C键长.同样地,N在石墨炔中替代环掺杂时形成的N-C2键与在链掺杂时形成的N-C1键长均为1.34 Å,小于N-C单键键长(1.38 Å[37]),并大于N-C双键键长(1.27 Å[38]).当N进行环掺杂时,N-C1键长大于N-C单键,而N进行链掺杂时,N-C2键长为1.18 Å,远小于N-C三键键长(1.22 Å)[37].B在石墨炔中进行环掺杂与链掺杂中时具有的Mulliken电荷分别为0.143 e和0.016 e,因此,B进行替代掺杂时会转移电子给碳原子,而且在环掺杂中的B转移更多电子.N在环掺杂中时获得0.247 e,而进行链掺杂时转移0.226 e.因此,当B,N原子分别在石墨炔中进行环掺杂和链掺杂时形成的结构将具有不同的电子性质.由文献可知,一般采用PBE计算得到的带隙会比实际偏小[39−42],但是能比较精确计算能带的Heyd−Scuseria−Ernzerhof方法特别耗时,因为主要是得到材料的导电趋势,不需要得到准确的带隙,因此从节约计算成本的角度,本文采用PBE方法计算了B和N原子分别在石墨炔中进行环掺杂和链掺杂时形成的结构的能带图(图2).由图2可知,四种掺杂结构的能带中均有能级穿过费米能级(Fermi energy,EF),因此,四种掺杂结构均为良好导体,体现了优越的电子传输性能.
为了进一步探索B,N原子掺杂的石墨炔的电子性质有什么具体不同之处,接下来继续计算了B,N及相邻C1,C2原子的分波态密度(partial density of states,PDOS),如图3所示.态密度图可以通过对体系的离散能级进行洛伦兹展开而得到,其展开系数为0.15 eV,由水平轨道给出的权重求和而得到.(EHOMO+ELUMO)/2被定义为费米能级,在图3中取在0 eV处,用黑色划线表示,其中EHOMO表示最高占据分子轨道,ELUMO表示最低未占据分子轨道.从PDOS图可以看出,当B,N原子在石墨炔中进行掺杂时,均有能态穿过费米能级,说明B,N掺杂使石墨炔从半导体变成导体.本文发现,B原子替代掺杂六元环上的C1原子时,B的p轨道与相邻C1,C2原子的p,s轨道在低能量区域(-6 - -4 eV)有重叠峰,同时,B原子与C2原子在远离费米能级的高能量区域也有重叠峰,因此,环掺杂的B原子与相邻C原子尤其是与C2原子之间存在更强烈的相互作用[43].而当B原子替代掺杂链上的C2原子时,B与C2原子之间有较强的重叠峰并穿过费米能级,相互作用没有环掺杂时强,表明B更倾向于替代C1原子,证实了以上的结论.同样地,当N原子替代掺杂链上的C2原子时,N与相邻C2原子的杂化有两个明显的重叠峰分布在费米能级两侧,而N替代掺杂六元环上的C1原子时则没有明显的重叠峰.这表明N更倾向于替代C2原子,与上文结论一致.

图2 四种结构的能带图 (a) B进行环掺杂;(b) B进行链掺杂;(c) N进行环掺杂;(d) N进行链掺杂Fig.2.Energy band of four structures: (a) B−ring doping;(b) B−chain doping;(c) N−ring doping;(d) N−chain doping.
由文献[44,45]可知,B,N原子在石墨炔中进行链掺杂和环掺杂时形成的四种结构的内聚能均接近实验上石墨的内聚能(7.37 eV),表明B,N原子掺杂的石墨炔结构稳定.由表1可知,B在石墨炔中进行环掺杂时的吸附能比链掺杂时低,而N掺杂恰恰相反.因此,B更倾向于掺杂在六元环上形成更稳定的结构,而N更倾向于掺杂在链上形成更稳定的结构.接下来对B原子在六元环上的掺杂稳定结构和N原子在链上的掺杂稳定结构进行300 K温度下的动力学模拟.本文使用Nose−Hoover C hain thermostats进行有限的温度调节,研究其热力学稳定性.计算发现,在经过步长为1 fs的5 ps模拟后,结构仍然非常完整,因此它们具有较好的热力学稳定性.

图3 不同掺杂位点B,N与相邻C1,C2原子的PDOS图Fig.3.The PDOS of B,N and neighboring C1,C2 atoms with different doping sites.
3.2 单个Li原子在B,N掺杂的石墨炔上的吸附
考虑到结构的稳定性及B,N原子在两处位置的结合能,选取B原子环掺杂的石墨炔和N原子链掺杂的石墨炔来进行储Li研究.为了包含完整的链和环,选用2 × 2 × 1的周期性晶胞来模拟单层结构.因为B和N原子分别比C原子少一个和多一个电荷,因此,首先计算了B,N掺杂结构带一个负电荷时的静电势来预测可能的储Li位点(图4).图4中红色代表电势高,绿色代表电势低,与未掺杂的纯碳石墨炔相同,大孔上方(H位点)和小孔上方(h位点)是Li合适的吸附位点.由图4可知,H位点比h位点电势更低.
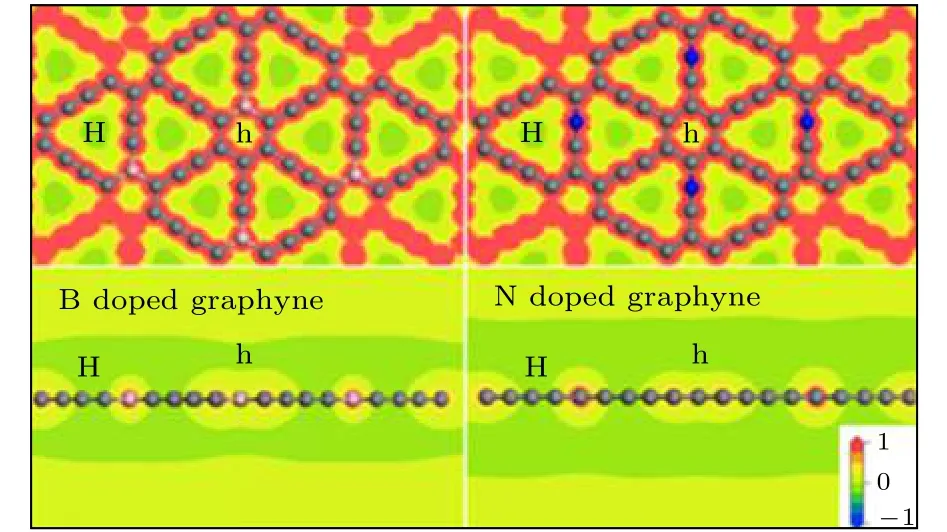
图4 B,N−掺杂石墨炔静电势的平视图与侧视图(静电势范围为1.0-1.0 Ha·e-1)Fig.4.Flat view and side view of the electrostatic poten−tial of B,N doped graphyne.The range of electrostatic po−tential is 1.0−-1.0 Ha·e-1.
通过以下公式来计算Li在B,N掺杂石墨炔上的平均吸附能,从而确定Li原子在B,N掺杂的石墨炔中的吸附强度[9]:

其中,ELi表示单个Li的总能量;EB/N−graphyne表示B,N掺杂石墨炔的总能量,而EB/N−graphyne+Li表示Li吸附的B,N掺杂石墨炔体系的总能量.计算发现,一个Li吸附在B掺杂的石墨炔的H位点和h位点的吸附能分别为4.16和3.27 eV,Li原子距离表面的吸附高度分别为0.87和1.83 Å.这和静电势图的预测很好地符合,H位点电势低,Li更倾向位于H位点.相比于在未掺杂石墨炔上的吸附能(H位点3.33 eV,h位点2.50 eV)[46],Li在B掺杂石墨炔上的吸附能大幅增加,这预示着B掺杂可以增强Li的结合.然而,当一个Li位于N掺杂石墨炔的H和h位置时,吸附能分别为2.75和2.32 eV,吸附高度分别为1.10和1.83 Å,相比于未掺杂石墨炔吸附能却有所降低.
为了阐明出现这种现象的原因,计算单个Li原子分别位于H和h位点时的电荷转移量以及差分电荷密度图.计算得知,Li位在B掺杂石墨炔的h位点时转移电荷为0.52 e,小于Li位于N掺杂石墨炔h位点时电荷转移量0.58 e.由于Li离子受到原子间斥力和静电作用力而能够稳定结合,但是B,N位于h位点时的吸附高度同为1.83 Å,因此B掺杂的石墨炔h位点处相比于N掺杂石墨炔h位点处的场强大、电势低,于是Li位于B掺杂石墨炔h位点时的吸附能大于N掺杂石墨炔.由图5(a)和图5(b)可以更清晰地看出,Li吸附于B掺杂石墨炔h位点时与B-C1键之间明显有电荷聚集,表明Li离子与B-C1键之间有较强的相互作用.当Li吸附于H位点时有相同的情况,有0.41 e电荷转移到B掺杂石墨炔,而有0.50 e的电荷转移到N掺杂石墨炔.在图5(c)中,Li离子与C2-C2三键之间有较多的电荷聚集,表明Li离子与B掺杂的石墨炔之间存在强烈的相互作用,而对于N掺杂石墨炔,Li离子仅与C2-C2三键之间有强烈的相互作用,与N-C2键之间的相互作用较弱.因此,N掺杂反而削弱了Li离子在石墨炔平面上的吸附强度.

图5 一个Li位于B掺杂的石墨炔(a) H和(c) h位点时的差分电荷密度图;一个Li位于N掺杂石墨炔(b) H和(d) h位−点的差分电荷密度图;其中差分电荷密度范围为-0.010.005 e/Å3,红色表示电子积聚,蓝色表示电子缺失Fig.5.Differential charge densities: One Li at (a) H and (c)h sites of the B−doped graphyne;one Li at (b) H and (d) h sites of the N doped graphyne.The range is -0.01−0.005 e/Å3,the red area stands for electron accumulation,and the blue area stands for electron deletion.
3.3 多个Li原子在B,N掺杂石墨炔上的吸附
接下来,将探索B,N掺杂的石墨炔的最大可逆储Li容量.众所周知,随着Li吸附量的增加,材料的结构可能发生剧烈变化.因此,在探索单个Li原子的稳定吸附位点的基础上,逐步增加单侧Li原子的数目,取最低能量结构以保证储Li结构的稳定性,并用以下公式计算Li的平均吸附能[9]:

其中,n是吸附的Li的总个数;ELi,EB/N−graphyne和EB/N−graphyne+nLi分别为单个Li原子的能量、B和N掺杂石墨炔的能量、Li和B及N掺杂石墨炔结构的总能量.将多个Li原子吸附结构命名位C11BLix和C11NLix.图6给出了多个Li原子在B,N掺杂石墨炔上的平均吸附能随x的变化规律.由图6可知,随着储Li量的增加,Li的平均吸附能明显降低,因为Li离子之间的排斥力使其难以吸附更多Li原子.N掺杂石墨炔的吸附能下降趋势比B掺杂石墨炔更为平缓,预示着N掺杂石墨炔有着更平缓的放电电压.
当B掺杂石墨炔单侧储Li量达到C11BLi11时,结构发生轻微形变,如图7(a)所示,此时Li原子的平均吸附能为2.15 eV.继续添加Li原子时,结构变形更加严重.由于负极材料较大的结构形变会导致较高的体积膨胀比,因此,B掺杂的石墨炔单侧最大储Li量为C11BLi11时对应的储Li容量为2061.62 mAh/g,几乎是未掺杂石墨炔的2.77倍[10]和石墨的5.54倍[47].对于N掺杂的石墨炔,当掺杂36个Li原子时即储Li容量达到C11NLi9时,结构发生轻微形变(图7(b)).Li的数量继续增加会导致更大的结构形变,当N掺杂石墨炔的最大储Li量为C11NLi9时,储Li容量高达1652.12 mAh/g.令人意外的是,虽然N掺杂降低了Li的吸附能,然而N掺杂石墨炔的储Li容量仍是未掺杂石墨炔的2.22倍.可以发现,当Li占据了最佳吸附位点后,后来的Li可以吸附到C2原子和N原子的顶位.这表明N掺杂虽然降低了Li的吸附能,但是增加了Li的可吸附位点,因此也能有效增加体系的储Li容量.

图6 多个Li在B,N掺杂墨炔上的平均吸附能随储Li数量的变化Fig.6.The Ead curves of multiple Li adsorbed on B,N doped graphyne.

图7 (a) B掺杂石墨炔最大Li结构的俯视图和侧视图;(b) N掺杂石墨炔最大储Li结构的俯视图和侧视图Fig.7.(a) Top and side view of the maximum Li adsorbed with B graphyne;(b) top and side view of the maximum storage Li adsorbed N doped graphyne.
另一个考量电极材料性能的重要参数是开路电压,由下列公式计算得到[32]:

其中,Ebcc−Li是Li的体心立方晶体的能量,e是电子的电荷量.图8给出了B,N掺杂石墨炔的开路电压随储Li容量的变化.Li离子电池为了获得高工作电压,负极的开路电压要尽可能低[48].此外,为了获得更好的工作性能,开路电压要尽可能平坦[49,50].从图8可以看出,在储Li容量很小的阶段,B掺杂石墨炔的开路电压达到2.32 V,随着储Li容量的增加,平均开路电压骤然下降到1.01 V.而N掺杂石墨炔的开路电压相对较低,平均开路电压为0.51 V,低于B掺杂石墨炔的平均值0.81 V.但是,值得注意的是,B掺杂石墨炔在高储Li容量阶段具有一段较为平坦的开路电压,容量为750-2061 mAh/g时的平均开路电压仅为0.42 V,小于未掺杂石墨炔的平均开路电压0.64 V[51].因此,B掺杂石墨烯具有更好的工作电压.
3.4 Li在B,N掺杂石墨炔上的扩散
为确定B,N掺杂石墨炔作为Li离子电池负极的充放电性能,计算Li在B,N掺杂石墨炔上的扩算能垒.如图9所示,Li在B,N掺杂石墨炔上均有3条不同的扩散路径,其中两条路径(path 1和path 2)为平面内扩散路径,一条路径(path 3)为面外扩散路径,垂直穿过平面到达另一侧.能垒大小可以通过过渡态搜索进行计算[52,53].

图8 B,N掺杂石墨炔的开路电压随储Li容量的变化,其中橙色划线表示B掺杂石墨炔的平均开路电压,紫色划线表示N掺杂石墨炔的平均开路电压Fig.8.Change curves of the open circuit voltage with the storage Li capacity for B,N doped graphyne.The orange dash line represents the average open circuit voltage of B doped graphyne,and the purple dash line represents the av−erage open circuit voltage of N doped graphyne.

图9 Li在B,N掺杂石墨炔上的扩散路径和对应的能量曲线图,图中红色曲线对应path 1上的扩散能垒;绿色曲线对应path 2上的扩散能垒;黑色曲线对应path 3上的扩散能垒Fig.9.Diffusion paths of Li on B,N doped graphyne and the corresponding energy curves.The red,green,black curves in the pan−els corresponds to the diffusion energy barrier on path 1,2,3,respectively.
计算发现,在B掺杂的石墨炔平面上,Li原子从H位点到相邻H位点的最小扩散能垒为0.82 eV,与未掺杂石墨炔的0.72 eV[54]相比增加了0.10 eV.Li原子从H位点到h位点的最小扩散能垒为0.99 eV,与未掺杂石墨炔的相比增加了0.07 eV.然而,当Li穿过大孔扩散到平面另一侧时,能垒仅为0.08 eV,低于未掺杂时的0.18 eV[54].这可以解释为,B掺杂拉长了键长,从而H位点处的孔径增大使Li更容易穿过平面.通过Li原子的扩散能垒计算发现,Li原子更倾向于在沿着远离B原子的方向上迁移.
与B掺杂石墨炔的情况恰好相反,N掺杂石墨炔面内Li原子的扩散能垒相比于未掺杂时大幅降低,Li原子从H位点到相邻H位点的最小扩散能垒仅为0.37 eV,接近Li在石墨烯上的扩散能垒0.32-0.48 eV[55,56].从H位点到h位点的最小扩散能垒为0.48 eV,降低了0.44 eV.但当Li穿过平面时,扩算能垒增加到0.34 eV,与未掺杂时相比增加了0.16 eV.恰好与B相反,N掺杂缩小了H位点处的孔径,因此Li穿过平面时的能垒增大了.同样,Li更倾向于在沿着远离N原子的路径上进行迁移.总体来说,B掺杂可以降低面外扩散能垒,增加面内扩散能垒;N掺杂的情况与其相反,N掺杂极大降低了石墨炔的面内扩散能垒,稍微增加了面外扩散能垒.因此,N掺杂可以明显提高Li的扩散性能.
4 结 论
本文使用DFT经过详细地深入研究发现经过B,N原子掺杂调控的二维多孔石墨炔在被用在Li离子电池负极材料时的储Li性能非常理想,主要得到如下3点结论.
1)通过比较B,N掺杂调控的二维多孔石墨炔的总能量发现B倾向于在石墨炔中进行环替代掺杂,而N倾向于在石墨炔中进行链替代掺杂.
2)通过计算Li在B,N原子掺杂调控的二维多孔石墨炔中的结合能得知,B掺杂可以有效增强Li与石墨炔之间的吸附作用,将可逆储Li容量增加到2061.62 mAh/g.N掺杂降低了Li原子与石墨炔之间的相互作用,但是增加了Li原子的可吸附位点,因此将可逆储Li容量提高到1652.12 mAh/g.
3)通过计算B,N掺杂调控的二维多孔石墨炔中Li的迁移势垒得知,B掺杂可以降低Li原子在垂直石墨炔平面的扩散能垒,对于面内扩散影响不大.N掺杂极大地提高了Li原子在石墨炔上的扩散性能,平面内能垒降至0.37 eV.
因此,B,N掺杂可从不同方面提升石墨炔作为Li电池负极材料的储Li性能.该研究可以为开发良好的储Li负极材料提供一个好的研究思路,为实验工作者提供理论依据.
猜你喜欢
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
燃料化学学报(2021年5期)2021-06-02
云南化工(2020年11期)2021-01-14
陶瓷学报(2020年3期)2020-10-27
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中国有色金属学报(2018年2期)2018-03-26
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
