
CO与H2在应变Fe(110)表面的竞争吸附*
2019-11-08 08:45李守英赵卫民乔建华王勇
物理学报 2019年21期
李守英 赵卫民 乔建华 王勇
1) (中国石油大学(华东)材料科学与工程学院,青岛 266580)
2) (青岛滨海学院机电工程学院,青岛 266555)
为研究CO降低临氢管线钢的氢脆机制,采用基于自旋极化密度泛函理论的第一性原理方法,研究H2和CO在Fe(110)表面吸附过程中的势能变化以及不同应变时的吸附.研究发现Fe(110)表面对CO的吸引力大于H2,且预先吸附的CO能阻碍H2的解离,减弱H与Fe之间的作用力.态密度分析结果表明CO中的C原子与Fe原子有多个共轭峰,有强烈的共轭杂化作用.不同应变Fe(110)表面的吸附结果表明CO在Fe(110)表面的吸附能比H2更负,CO与表面的结合强度更大,CO优先吸附.结合热力学定量计算分析CO分压对氢覆盖度影响,结果表明随着CO的分压升高,氢覆盖度降低.表面拉应变越大,需要的CO分压越高.拉应变使得H2,CO吸附能差减小,CO阻碍氢吸附的能力降低是拉应变表面需要更高CO分压的原因.
1 引 言
近年来,环境保护、地球温室效应、能源枯竭已经成为世界各国共同努力研究的重要课题,各国都在致力于开发清洁能源和可再生能源[1].太阳能、风能具有丰富、清洁、可再生的优点而受到广泛关注.但是这些可再生能源具有间歇性、地域性、且不易储存和运输的特点[2,3].相对而言,氢具有高效、可存储和运输的优点,被视为最理想的能源载体.世界各国致力于研究将太阳能、风能产生的电力转换为氢能[4,5].大量、长距离氢能输送,管道是最高效、低成本的方式[6,7].管线钢在输送氢气的过程中管道内的高压氢环境会引起氢的吸附、渗透,使得氢进入管线钢内部,进而导致管线钢的氢致脆化[8,9].氢脆是影响输氢管线工作最突出的因素.焊接接头区域由于表面存在焊接残余应力及工作应力是输氢管线的薄弱环节[10].表面应力改变材料的表面电子结构,影响吸附[11−13].氢的解离吸附是氢进入管线钢内部的前提.目前铁表面的氢吸附主要围绕无应力表面的吸附[14−16],关于应力加载铁表面的氢吸附未见报道.
当前,CO和H2在金属Ni,Fe等表面混合吸附研究目的主要是催化合成碳氢化合物[17,18],降低钢的氢脆的研究较少.Bernasek等[19]通过TEAS(thermal energy atom scattering)试验研究高真空度下CO和H2在Fe(111)面的吸附,发现预先饱和吸附CO后,表面不再吸附H2.Wang等[20]研究发现在Fe(111)面,每个CO分子能阻碍2个H原子的吸附.Huo和Liao[18]研究发现CO与H2的稳定吸附构型与表面CO/H2覆盖度比值有关.通过比较吸附能,多数学者们认为CO分子优先于H2在铁表面吸附[21,22].但是关于CO对H2的解离过程以及CO分压对吸附氢覆盖度的影响鲜有报道.
Fe(110)面是α−Fe的密排面,具有最低的表面能,是塑性变形过程中的新增台阶面[23,24].本文基于密度泛函理论,研究在应变Fe(110)表面,CO对氢解离、吸附以及覆盖度的影响.本文研究结果对于CO定量掺杂减少氢吸附、降低临氢管线氢脆具有重要的理论指导意义.
2 计算模型和方法
本文计算应用Material Studio软件中基于密度泛函理论的CASTEP[25]模块完成,电子波函数采用平面波展开,布里渊区采用Monkhorst−Pack方法均匀k点取样,应用广义梯度近似的rPBE[26]泛函(修正后的Perdew−Burke−Ernzerhof泛函)描述电子之间的交换关联能,利用超软赝势[27]描述价电子与离子实之间的关系,选取平面波截断能为425 eV,自洽场循环收敛于1.0 × 10-5,力收敛于0.03 eV/atom,几何优化采用Broyden−Fletcher−Goldfarb−Shanno算法.α−Fe单胞优化k点取19 × 19 × 19,优化后的晶胞参数为a=2.845 Å,与实验结果2.866 Å基本一致.基于优化后的单胞模型,构建7层2 × 2的Fe(110)层模型,设置真空层厚度为12 Å.表面模型以及吸附模型几何结构优化时,k点取值为10 × 10 × 1,底下4层Fe原子固定,上面3层Fe原子以及吸附原子进行弛豫.文中所选参数均进行了收敛性测试.先后将CO与H2分子放入12 Å × 12 Å × 12 Å的立方盒子进行优化,k点取1 × 1 × 1,优化后CO分子的键长为1.154 Å,H2键长为0.748 Å,分别与实验值1.128 Å,0.741 Å符合得较好.
Fe(110)表面对称点,如图1所示.top表示顶位,lb表示长桥位,sb表示短桥位,tf表示三重洞位.

图1 Fe(110)面及其对称点Fig.1.Fe(110) surface and high symmetry sites.

式中l0表示无应变的晶胞模型尺寸,Δl表示该尺寸的变化量.应变施加范围为-2%-+2%.
自由气体分子i在表面吸附过程中吉布斯自由能的变化通过(2)式求解:

H2与CO的混合吸附过程中,氢的覆盖度θH通过(3)式[29]计算:

3 计算结果与讨论
3.1 CO分子的吸附
CO分子的C原子容易与金属原子作用[30].在CO吸附计算中,C原子靠近表面的对称点top,sb,lb,tf.几何结构优化完成后,发现CO垂直吸附于上述对称点.用(4)式定义CO的吸附能:

式中,Eslab,Eslab+CO分别表示Fe(110)吸附CO分子前后表面的能量;ECO表示CO分子能量;表示CO分子的吸附能.
由吸附能的定义式可知,负值表示吸附过程放出热量,正值表示吸收热量.吸附能的值越负,表示该吸附构型越稳定.CO以及H2在不同位置的吸附能如表1所列.从表1数据可以看出本文计算结果与文献[31]数据符合得很好,误差仅在0.5%,说明本文计算结果是可信的.微小的数据差别可能来自于rPBE与PBE近似的不同.由表1数据可以看出,4个位置CO的吸附能都为负值,且top位的吸附能值最负,表明该位置是CO分子最优最稳定吸附位置.低能电子衍射试验也表明CO分子垂直吸附在top位[32].

表1 CO与H2在Fe(110)表面不同位置的吸附能(eV)Table 1.Adsorption energies of CO and H2 on high symmetry sites of Fe(110).
图2为CO分子吸附在top位前后的分波态密度(PDOS)图.可以看出吸附前,孤立的CO分子中C原子和O原子有强烈键合作用,从低能到高能的分子轨道分别是3σ,4σ,1π,5σ,2π.其中5σ是最高占据分子轨道,2π是最低未占据分子轨道.吸附前,Fe原子的态密度分布在-8-22 eV能量区间.CO吸附使得Fe原子自旋向上态密度和自旋向下态密度差异减小.吸附后,Fe原子的4s,3p在-22.6 eV,-8.9 eV处,3d在-5.7 eV出现较大的共轭峰.表明表面Fe原子与C原子存在强烈的杂化耦合作用.该杂化耦合作用使得C原子的2p态展宽,形成成键态和反键态.成键态使得C原子与Fe原子的作用增强,是CO在Fe表面吸附能较大的原因.
3.2 H2分子的吸附
计算过程中发现,在靠近Fe(110)表面时,氢分子容易解离成为氢原子吸附在Fe(110)表面.为提高计算效率,通过氢原子的吸附根据(5)式计算氢分子的吸附能

式中Eslab,Eslab+H分别是表面吸附氢原子前后体系的能量;EH2表示氢分子的能量.表1列出了氢分子的吸附能.
氢分子在tf位置的吸附能最负,为-1.33 eV,表明tf位的吸附最稳定,与Wang等[20]的计算数据-1.36 eV基本一致.比较CO和H2的吸附能数据可以看出,4个吸附位CO的吸附能均比H2的吸附能更负,表明CO与Fe(110)表面的结合强度大于H2与Fe(110)面的结合强度.其中CO吸附在sb位与H2吸附在tf之间结合强度差值最小,为0.31 eV.
计算了H原子在表面tf位吸附前后的态密度,如图3所示.从图3可以看出,吸附后H的1s电子向左移动到-7.3- -5.2 eV,存在明显的波峰.Fe原子的4s电子在-7.8- -5.8 eV之间出现新的峰、3p电子与3d电子在-6.2 eV出现弱小的峰,表明Fe的4s,3p,3d电子与H的1s电子存在共轭杂化作用,共轭峰值较小,说明杂化作用较弱.H原子的吸附对Fe原子自旋向上和自旋向下的态密度差异影响较小.

图2 CO,Fe(110)面及其top位吸附前后分波态密度图 (a)吸附前;(b)吸附后Fig.2.Projected local density of states from Fe on clean Fe surface and a CO molecular in vacuum (a) and with a CO adsorbed surface (b).

图3 H与Fe(110)面tf位吸附前后分波态密度图 (a)吸附前;(b)吸附后Fig.3.Projected local density of states from Fe on clean Fe surface and a H atom in vacuum (a) and with a H adsorbed surface (b).
3.3 势能随距离的变化
为分析CO对H2的吸附以及解离过程的影响,计算H2,CO分子逐渐靠近Fe(110)表面的势能变化.计算过程中限定吸附物的Z向坐标,仅对X,Y坐标进行弛豫.
Wang 等[33]研究发现垂直构型靠近铁表面的H2难以解离.且前面分析表明CO垂直吸附于Fe(110)表面.因此本文分别计算与表面垂直的CO、平行H2以及预先吸附CO平行的H2在靠近表面过程中的势能变化,计算结果如图4所示.图中横坐标各距离分别为CO中的C原子到表面的距离、H2两个H原子的中心到表面的距离以及预先吸附CO时H2的中心到表面的距离.H2在靠近表面的过程中,能量先降低,后又逐渐升高,到2.4 Å能量到达最高点.结构优化中发现距离小于2.4 Å时,如不固定H2的Z坐标,H2直接解离为H原子并吸附在tf位.距离减小过程中,CO的吸附能不断降低,到达1.5 Å时,能量最低,形成CO的分子吸附态.能量随距离变化的斜率表示作用力的大小.1.5-3 Å范围内,表面对CO的吸引力明显大于H2,这是表面优先吸附CO的原因之一.
Gholizadeh和Yu[34]研究认为小分子的解离过程需要考虑零点能校正.为准确计算H2的解离能,同时提高计算效率,通过频率计算对吸附H2,CO+H2的初始结构以及其在2.4 Å的结构进行零点能校正.零点能校正公式如(6)式:

式中,vj表示振动频率,h表示普朗克常数.

图4 CO与H2吸附过程中的势能变化Fig.4.Potential energy variations of CO and H2 moving to−wards Fe(110).
经零点能校正后,H2的解离能为0.08 eV,与文献[35]的0.1 eV基本一致,表明H2分子易解离.预先吸附CO后,H2的解离能升高到0.13 eV,说明预先吸附CO阻碍H2的解离,并且使得最终吸附在tf位的H2的吸附能由-1.33 eV升高-1.10 eV,表明预先吸附的CO使得H和Fe(110)面的作用减弱.
3.4 应变对CO与H2吸附的影响

图5 CO吸附能、H2吸附能和应变之间的关系Fig.5.Relationship between adsorption energy of CO and H2 and strain.

图6 H2和CO吸附能的差与应变之间的关系Fig.6.Relationship between strain and the difference of CO and H2 adsorption energy.
计算了应变-2%-2%之间Fe(110)面的CO吸附.发现应变表面sb,lb,tf位置的CO均向top位弛豫,最终吸附在Fe(110)的top位置.top位CO的吸附能以及tf位H2的吸附能和应变之间的关系如图5所示,两者之间的吸附能差如图6所示.可以看出H2的吸附能与应变之间存在近似负的线性关系.拉应变使得H2的吸附能更负,压应变使得吸附能值升高.这表明拉应力使得氢与Fe(110)面的作用力增强,压应力减小了氢与Fe(110)面的相互作用.与H2的吸附相反,拉应变使得CO吸附能升高,吸附作用减弱;压应变使得吸附能更低,吸附作用增强.在计算应变范围以内,CO的吸附能均比H2的吸附能更负,说明CO与Fe的作用力更强.通过图6可以看出,拉应变使得H2与CO的吸附能差ΔE减小,压应变使得ΔE增大.压应变使得CO阻碍氢吸附的效果更为明显.
为定量分析CO对应变Fe(110)面氢覆盖度θH的影响,将CO,H2的吸附能代入(2)式,利用(2)式计算出的吉布斯自由能的变化代入(3)式计算298 K时的θH.图7表示298 K,10 MPa的H2分压条件下,CO的分压与θH之间的关系.从图7可以看出,无论表面有无应力,随着CO的分压升高,氢覆盖度降低.CO分压升高到一定值时,氢覆盖度接近0,表明CO掺杂能抑制氢吸附.-2%,0,2%平面应变Fe(110)表面,θH降低到1%时,CO分压分别为105 Pa,1.1 × 103Pa,2.4 ×105Pa.拉应变使得H2和CO吸附能差值减小,CO阻碍氢吸附的能力降低,需要更高的CO分压降低氢的覆盖度.
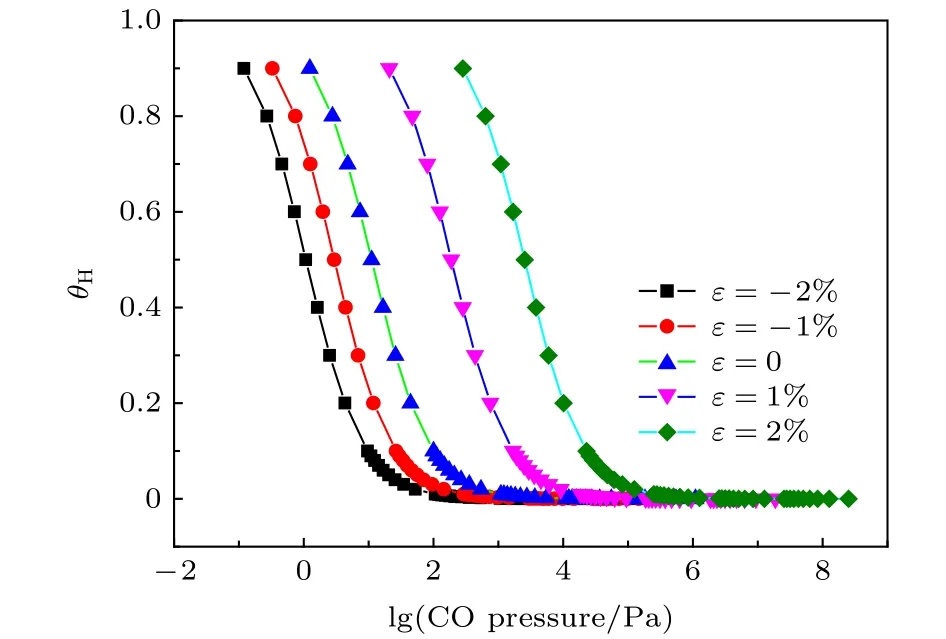
图7 CO的分压与θH之间的关系Fig.7.Relationship between CO pressure and coverage of H.
4 结 论
本文采用密度泛函理论研究H2,CO在应变Fe(110)表面的竞争吸附特性.主要得出如下结论:
1)预先吸附CO阻碍H2的解离,使得H2与Fe(110)面的作用减弱;
2)态密度分析结果表明CO中的C原子与Fe原子存在强烈的杂化耦合作用,而H原子与Fe原子的杂化作用较弱;
3)无论表面有无应变,CO与表面的结合强度均高于H2;拉应变使得两者吸附能差减小,CO阻碍氢吸附的能力降低;
4) CO掺杂对氢覆盖度的计算发现,随着CO的分压升高,氢覆盖度降低;拉应变越大,CO阻碍氢吸附的作用越弱,需要的CO分压越高;2%平面应变Fe(110)表面,θH降低到1%时,CO分压为2.4 × 105Pa.
感谢中国石油大学张宏玉老师在计算过程中给予的指导和帮助.
猜你喜欢
科学技术创新(2022年30期)2022-10-21
无机化学学报(2022年9期)2022-09-16
农业与技术(2021年23期)2021-12-14
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
黑龙江水利科技(2020年8期)2021-01-21
原子与分子物理学报(2020年5期)2020-03-17
中国塑料(2016年1期)2016-05-17
读写算·教研版(2016年8期)2016-05-07
