
New insights into Wnt signaling alterations in amyotrophic lateral sclerosis: a potential therapeutic target?
2020-03-07 05:31CarlosGonzlezFernndezPauGonzlezFranciscoJavierRodrguez
中国神经再生研究(英文版) 2020年9期
Carlos González-Fernández, Pau González, Francisco Javier Rodríguez
Laboratory of Molecular Neurology, Hospital Nacional de Parapléjicos (HNP), Toledo, Spain
Abstract Amyotrophic lateral sclerosis is a fatal neurodegenerative disorder characterized by upper and lower motor neuron degeneration, which leads to progressive paralysis of skeletal muscles and, ultimately, respiratory failure between 2-5 years after symptom onset. Unfortunately, currently accepted treatments for amyotrophic lateral sclerosis are extremely scarce and only provide modest benef it. As a consequence, a great effort is being done by the scientific community in order to achieve a better understanding of the different molecular and cellular processes that inf luence the progression and/or outcome of this neuropathological condition and, therefore, unravel new potential targets for therapeutic intervention. Interestingly, a growing number of experimental evidences have recently shown that, besides its well-known physiological roles in the developing and adult central nervous system, the Wnt family of proteins is involved in different neuropathological conditions, including amyotrophic lateral sclerosis. These proteins are able to modulate, at least, three different signaling pathways, usually known as canonical (β-catenin dependent) and non-canonical (β-catenin independent) signaling pathways. In the present review, we aim to provide a general overview of the current knowledge that supports the relationship between the Wnt family of proteins and its associated signaling pathways and amyotrophic lateral sclerosis pathology, as well as their possible mechanisms of action. Altogether, the currently available knowledge suggests that Wnt signaling modulation might be a promising therapeutic approach to ameliorate the histopathological and functional deficits associated to amyotrophic lateral sclerosis , and thus improve the progression and outcome of this neuropathology.
Key Words: ALS; astrocytes; Frizzled; human; microglia; motor neuron; neurodegeneration; neuroinf lammation; spinal cord; Wnt
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative condition characterized by both upper and lower motor neuron (MN) degeneration, progressive paralysis of skeletal muscles and respiratory failure within 2-5 years of symptom onset (Riva et al., 2016; Oskarsson et al., 2018). The global incidence of ALS is around 2-3 cases per 100,000 persons annually in the general adult population, although it has been described that risk increases with age with a peak around 75 years, and that its prevalence is higher in men than in women with a ratio of about 1.5 (Al-Chalabi and Hardiman, 2013; Morgan and Orrell, 2016; Oskarsson et al., 2018). Approximately 90-95% of ALS cases are defined as sporadic (sALS), meaning that there is not a clear genetic inherited component and the disease seems to occur randomly, while approximately 5-10% of ALS cases are related with several heritable genetic mutations and thus are classif ied as familial (fALS) (Morgan and Orrell, 2016).
The most common known genetic cause of ALS, also associated with frontotemporal dementia, is the C9orf72 repeat expansion, accounting for 30-40% of fALS and a small fraction of sALS (Ji et al., 2017; Oskarsson et al., 2018). Together with C9orf72, other mutations such as that observed in copper-zinc superoxide dismutase (SOD1), transactive response DNA-binding protein 43 and fused in sarcoma genes comprise more than 50% of fALS cases (Morgan and Orrell, 2016; Oskarsson et al., 2018). Nevertheless, the development of ALS seems to be a multistep process in which gene mutations depict only one of the steps that trigger the disease. Indeed, it has also been described that several environmental factors may be related to an increased risk of ALS, such as smoking, physical activity, pesticide exposure or trauma, among others (Al-Chalabi and Hardiman, 2013; Gordon, 2013).
The variety of genetic mutations identif ied in fALS cases have led to the development of different transgenic animal models, which show similar pathological events to those observed in patients. Among them, mouse models are the most broadly used, providing the most closely translatable results to humans (Mancuso and Navarro, 2015). At this point, since most studies have been performed in animal models of fALS, it should be noted that both sALS and fALS are clinically and pathologically similar (Tan et al., 2017) and, therefore, that a better understanding of the harmful processes underlying fALS may provide a greater comprehension of the neurodegenerative mechanisms in sALS.
From a pathophysiological point of view, ALS is likely to be the consequence of different dysregulated processes including aberrant RNA metabolism, glutamate-mediated excitotoxicity, impaired protein homeostasis, defective axonal transport, oxidative stress, mitochondrial dysfunction, neuroinflammation and activation and proliferation of astroglial and microglial cells, which ultimately lead to neuronal degeneration (Mancuso and Navarro, 2015). However, the pathological mechanisms of ALS are still far away from being well understood. Moreover, several drugs proven to be efficient in animal models of ALS have failed in clinical trials and, to date, the main therapy available for ALS treatment is the glutamate antagonist Riluzole, which has substantial adverse side effects and only provide modest benef it, since it only prolongs patient survival about 3 months (Lacomblez et al., 1996; Cheah et al., 2010). More recently, a free radical scavenger known as Edaravone has been also approved for the treatment of ALS, since it slows disease progression, albeit only in a small subset of patients (Oskarsson et al., 2018).
In this complex scenario, the identification of other genes and molecular pathways responsible or involved in ALS is of utmost importance to shed light on the pathological mechanisms of the disease leading to MN degeneration and thus unravel new potential targets for therapeutic intervention. In this sense and as it will be discussed in detail, over the last years an increasing number of experimental studies have clearly pointed to the potential involvement of the Wnt family of proteins and its associated signaling pathways in this neuropathological condition. To this end, we conducted an electronic search on PubMed which included all articles that were published up to 2019 and with the keywords: Wnt [and] ALS, Wnt signaling [and] ALS, Frizzled [and] ALS.
Wnt Signaling Overview
The Wnt family of proteins is one of the most evolutionarily conserved regulators of crucial aspects in embryo development and adult tissue homeostasis (Logan and Nusse, 2004; Ille and Sommer, 2005; Zhang et al., 2011; Salinas, 2012). This family of proteins is composed by 19 secreted cysteine-rich glycoproteins (Wnt1, Wnt2, Wnt2b, Wnt3, Wnt3a, Wnt4, Wnt5a, Wnt5b, Wnt6, Wnt7a, Wnt7b, Wnt8a, Wnt8b, Wnt9a, Wnt9b, Wnt10a, Wnt10b, Wnt11, and Wnt16), 10 seven-pass transmembrane Frizzled receptors (Fz1-10), 2 low-density lipid receptor-related proteins (LRP5/6) co-receptors, and other non-conventional receptors such as tyrosine kinase-like orphan receptors (ROR1/2), protein tyrosine kinase 7 and the receptor-like tyrosine kinase (Ryk). Moreover, Wnt signaling is regulated by multiple secreted modulators, including the Dickkopf proteins (Dkk1-4), the secreted frizzled-related proteins (sFRPs1-5) and the Wnt inhibitory factor 1 (Wif1) (Hendrickx and Leyns, 2008; Niehrs, 2012).
Several divergent intracellular signaling pathways are activated depending on the Wnt-receptor combination. The best characterized is the so-called “canonical” or “β-catenindependent” signaling pathway, in which β-catenin protein is the main mediator. Brief ly, in the absence of Wnt signal, cytosolic β-catenin levels are maintained low through the action of a ubiquitin-dependent multiprotein destruction complex composed by the scaffolding proteins Axin and adenomatous polyposis coli, and the kinases responsible for the β-catenin phosphorylation casein kinase 1 (CK1α) and glycogen synthase kinase 3β (GSK-3β) (Figure 1A). The binding of the Wnt ligand to the corresponding Fz receptor and LRP5/6 co-receptor triggers the activation of the Dishevelled (DVL) protein, leading to the inhibition of the destruction complex and the subsequent accumulation and translocation of β-catenin to the nucleus, where it can act as a co-activator together with members of the T cell factor family (Tcf1-4) and the lymphoid enhancer factor 1 (Figure 1A) (Michaelidis and Lie, 2008; Zimmerli et al., 2017; Steinhart and Angers, 2018).
In addition to the canonical Wnt signaling, there are two alternative non-canonical β-catenin independent pathways known as the “Wnt/Ca2+pathway” and the “Wnt/planar cell polarity pathway” (Wnt/PCP) (Michaelidis and Lie, 2008; Kahn, 2014; Lambert et al., 2015). In the Wnt/Ca2+pathway, the binding of Wnt ligands to their specific receptors promotes the release of intracellular Ca2+and the subsequent activation of Ca2+-dependent proteins, such as protein kinase C (PKC) and calcium-calmodulin-dependent protein kinase II (CaMKII) (Figure 1B). Finally, these proteins regulate the nuclear transcription factor of activated T-cells (NF-AT). In the Wnt/PCP pathway, the interaction of the Wnt ligand with the corresponding receptor triggers a signaling cascade that involves small GTPase proteins, such as RhoA, Rac1 and Cdc42, which eventually activate the c-Jun N-terminal kinase (Figure 1C).
Adding further complexity to this classical view of the Wnt signaling pathways, it has been found that they are highly connected, establishing a Wnt signaling network in which canonical and non-canonical pathways often overlap to coordinate complex cellular responses. Indeed, canonical and non-canonicalWnt signaling pathways crosstalk and antagonize each other. Moreover, it has been observed that some Wnt ligands are involved in more than one of these signaling pathways depending on the receptor and co-receptors that are activated in each specif ic condition and, therefore, that the same Wnt ligand is able to regulate different biological processes that were originally assigned to opposite Wnt pathways (Topol et al., 2003; Kestler and Kuhl, 2008). Owing to the great number of Wnt ligands, receptors, co-receptors and modulators, as well as to the high complexity of the Wnt-associated signaling pathways, the Wnt family of proteins is able to modulate a wide range of cellular processes, such as cell fate determination, cellular polarity, cell contractility and cytoskeleton remodeling, cell proliferation, cell cycle arrest and differentiation, cell migration and invasion, cell survival and convergent extension and axon guidance processes, in different tissues and organs under both physiological and pathological conditions (Michaelidis and Lie, 2008; Clark et al., 2012; Niehrs, 2012; Ring et al., 2014; Sebbagh and Borg, 2014; Lambert et al., 2015; Xiao et al., 2017).

Figure 1 Schematic overview of Wnt signaling pathways.
Interestingly, a growing number of studies are showing that Wnt-related molecules play a major role in different cellular and molecular processes that characterize the progression and outcome of various central nervous system pathologies, such as neuron survival (Alvarez et al., 2004; Caricasole et al., 2005; Inestrosa and Toledo, 2008; Liu et al., 2008a; Toledo et al., 2008; Mastroiacovo et al., 2009), astroglial and microglia/macrophage reactivity and inf lammation (Lutgen et al., 2016; Yang et al., 2016; Ding et al., 2017; Gonzalez and Rodriguez, 2017), axonal regeneration and/or degeneration (Liu et al., 2008b; Miyashita et al., 2009; Suh et al., 2011; Hollis and Zou, 2012; Rodriguez et al., 2014), blood-brain barrier dysfunction (Jean LeBlanc et al., 2019) and/or mobilization and differentiation of neural precursors (Parish et al., 2008; Yin et al., 2008; Cui et al., 2011; Gonzalez-Fernandez et al., 2016a). Accordingly, Wnt signaling alterations have been associated with several central nervous system disorders including Alzheimer’s (Caricasole et al., 2004; Inestrosa and Toledo, 2008; Tapia-Rojas and Inestrosa, 2017, 2018), Huntington’s (Wei et al., 2001; Godin et al., 2010) and Parkinson’s (Parish et al., 2008; L’Episcopo et al., 2011b, 2018) diseases, multiple sclerosis (Yuan et al., 2012; Xie et al., 2014; Vallee et al., 2018) and, more recently, ALS (Chen et al., 2012a, b; Li et al., 2013; Pinto et al., 2013; Wang et al., 2013; Yu et al., 2013; McLoon et al., 2014; Tury et al., 2014; Gonzalez-Fernandez et al., 2016b, 2019; Ouali Alami et al., 2018).
Wnt Alterations in Experimental Models of Amyotrophic Lateral Sclerosis
To date, the bulk of knowledge about the potential contribution of Wnt signaling disturbances to the ALS pathogenesis comes from some in vitro studies and from research conducted in the SOD1G93Atransgenic mouse model of ALS (Chen et al., 2012a, b, 2017; Li et al., 2013; Pinto et al., 2013; Wang et al., 2013; Yu et al., 2013; Tury et al., 2014; Gonzalez-Fernandez et al., 2016b; Bhinge et al., 2017; Ouali Alami et al., 2018). These mice, which overexpress the mutant SOD1 human protein, reproduce most of the clinical and neuropathological findings of human ALS and play a critical role in the ALS research field, representing the most widely employed in vivo ALS model so far.
The first evidences identif ied in this experimental model of ALS, pointing to a potential role of the Wnt family of proteins in this neuropathology, showed that both mRNA and protein levels of different Wnt family members were evidently altered in the spinal cord of ALS transgenic mice compared with wild-type (WT) littermates. More specif ically, a transcriptional microarray study to investigate the mRNA expression of most of the Wnt signaling components and target genes in the spinal cord revealed that many of them showed an altered expression in the ALS transgenic mice at specif ic stages of the disease (Yu et al., 2013), providing the first clear evidence of a global Wnt signaling dysregulation associated with this neuropathology. Brief ly, the authors observed that the expression of most Wnt ligands was significantly upregulated in this neuropathological condition (Wnt1, Wnt2, Wnt3, Wnt3a, Wnt4, Wnt5a, Wnt5b, Wnt7a, Wnt7b, Wnt8a, Wnt9a, Wnt10a, Wnt11 and Wnt16), while only the expression of Wnt8b and Wnt16 was found to be downregulated. Moreover, the expression of all Fz receptors and LRP5 co-receptor was also upregulated in the spinal cord of ALS transgenic mice. Likewise, some Wnt secreted modulators were also strongly upregulated (Wif1, sFRP3, sFRP4), while others were found to be downregulated (sFRP1, sFRP3). In agreement, several studies have shown that the mRNA levels of specif ic Wnt-related members (Wnt1, Wnt2, Wnt3a, Wnt5a, Wnt7a, Fz1, Fz2) were found to be upregulated in the spinal cord of ALS transgenic mice (Chen et al., 2012a, b; Li et al., 2013; Wang et al., 2013). In this line and further supporting that the expression of the Wnt family of proteins is evidently altered in this neuropathological condition, we have recently published a study showing that the mRNA expression of a wide range of Wnt-related molecules was found to be either upregulated (Wnt5a, Wnt7b, Fz1, Fz2, Fz3, Fz8, ROR2 and Wif1) or downregulated (Wnt4 and Fz4) in the spinal cord of ALS transgenic mice (Gonzalez-Fernandez et al., 2016b).
Furthermore, some of these studies have also evaluated whether the previously detailed variations in the gene expression of different components of the Wnt family of proteins led to similar changes in their protein expression. Accordingly to that observed at mRNA level, the protein expression of Wif1 and Wnt4 (Yu et al., 2013), Wnt2 and Wnt7a (Chen et al., 2012b), Wnt5a and Fz2 (Li et al., 2013), Wnt1 (Wang et al., 2013) and Wnt3a (Chen et al., 2012a) was also upregulated in the spinal cord of the ALS transgenic mice whereas, surprisingly, Fz1 protein levels were decreased (Wang et al., 2013), suggesting the potential involvement of post-translational regulatory mechanisms in this particular case. Interestingly, most of these studies did not only find alterations in the global protein levels of the previously detailed Wnt-related molecules, but also in their spatial and cellular distribution, mainly in astroglial and neuronal cells. More specif ically we and others have shown that, under physiological conditions, Wnt4 and Wif1 (Yu et al., 2013), Wnt2 and Wnt7a (Chen et al., 2012b), Wnt1 and Fz1 (Wang et al., 2013), Wnt5a and Fz2 (Li et al., 2013), Wnt3a (Chen et al., 2012a) and Fz5 (Gonzalez-Fernandez et al., 2016b) were expressed mainly in neurons and, to a lesser extent, in astrocytes, while the non-canonical Ryk receptor was found to be expressed in MNs and oligodendrocytes, but not in astrocytes, in the ventral horn of the lumbar spinal cord (Tury et al., 2014). In contrast, in the spinal cord of the ALS transgenic mice the amount of Wnt1, Wnt2, Wnt3a, Wnt4, Wnt5a, Wnt7a, Fz1, Fz2 and Wif1, but not Fz5-expressing astrocytes was notably increased in the locus of degeneration at the ventral horns of the spinal cord (Chen et al., 2012a, b; Li et al., 2013; Wang et al., 2013; Yu et al., 2013; Gonzalez-Fernandez et al., 2016b). Accordingly, Wnt1 (Wang et al., 2013), Wnt5a and Fz2 (Li et al., 2013) protein levels were found to be upregulated in primary cultured astrocytes from ALS mice spinal cords. At the neuronal level, we recently described that Fz5 immunoreactivity showed a marked increase throughout ALS progression in some neuronal cells located in the ventral horns that might be undergoing degenerative processes (Gonzalez-Fernandez et al., 2016a). Moreover, it has been shown that the expression of the non-canonical Ryk receptor was increased in MNs and ventral white matter axons in the spinal cord of ALS mice (Tury et al., 2014). On the contrary, the immunoreactivity of other Wnt-related molecules such as Wnt3a, Wnt4, Wnt5a or Fz2 in neuronal cells was significantly reduced in the ventral horns of the ALS mice spinal cord, possibly due to the loss of neurons associated with this pathology (Chen et al., 2012a; Li et al., 2013; Yu et al., 2013; Gonzalez-Fernandez et al., 2016a). Finally, some in vitro studies also described that Wnt1 (Wang et al., 2013) as well as Wnt5a and Fz2 (Li et al., 2013) proteins expression were downregulated in differentiated neuroblastoma N2a cells and MN-like NSC34 cells expressing mutated hSOD1, respectively, which might be related to the MN death process in ALS.
Besides, it has also been shown that the previously detailed alterations in the spatio-temporal and cellular expression pattern of different Wnt ligands, receptors and modulators led to variations in the activation state of different Wnt-related signaling pathways in this neuropathological condition. Regarding the canonical Wnt/β-catenin signaling pathway and in agreement with the above-mentioned upregulation of prototypical canonical Wnt ligands, receptors and co-receptors, different studies have pointed to the activation of this signaling pathway in the ALS transgenic mice. For instance, although no changes were observed in the expression of total GSK-3β, inactive phospho-GSK-3β protein level was significantly upregulated in the ALS transgenic mice, pointing to the activation of the canonical Wnt signaling pathway (Chen et al., 2012b). In agreement, although no variations were found in total β-catenin mRNA and protein levels between WT and ALS mice in the spinal cord, an increase of nuclear β-catenin was observed in the spinal cord during the progression of the disease (Chen et al., 2012a). Finally, with several exceptions, the expression of different target genes of this signaling pathway such as Cyclin D1, Myc, Sox 17 or Fosl1, was upregulated mainly at the final stage of the disease (Yu et al., 2013). Regarding the cellular types where the canonical Wnt/β-catenin signaling pathway was activated in the ALS mice spinal cord, it is interesting to note that, under physiological conditions, β-catenin was mainly detected in neurons, and in a small amount of astrocytes (Chen et al., 2012a). In contrast, β-catenin expressing neurons were decreased, possibly due to the neuronal loss processes that take place during the progression of ALS, while β-catenin+astrocytes were found to be increased in the spinal cord of ALS transgenic mice, strongly suggesting that the activation of the Wnt/β-catenin signaling pathway observed in this neuropathological condition mainly occurs in astroglial cells. In agreement, the expression of Cyclin D1, which is a well-known target gene of this signaling pathway, is robustly increased in astroglial cells in the ALS mice spinal cord (Chen et al., 2012a). Moreover and in agreement with the previously detailed ALS-related decrease in the presence of β-catenin expressing neurons, an in vitro study, where the endogenous activity of the canonical Wnt/β-catenin pathway was analyzed in the MN-like NSC34 cell line expressing WT or G93A mutated forms of human SOD1, showed that transcriptional activity of β-catenin was reduced in mutated hSOD1 expressing cells concomitant to the cytosolic aggregation of β-catenin, which probably alters its nuclear translocation and reduces its transcriptional function (Pinto et al., 2013; Pinto et al., 2019). Nevertheless, some discrepancies have been found in other in vitro studies, in which differentiated MNs from mutant SOD1 patient-derived iPSCs showed an activation of the canonical Wnt/β-catenin signaling pathway, as the authors found an increased nuclear localization of β-catenin compared with control and isogenic corrected MNs (Bhinge et al., 2017). On the other hand, although our current knowledge about the potential alterations in the activation state of non-canonical Wnt signaling pathways in the pathology of ALS is scarcer, it should be noted that evident variations in the mRNA and protein expression of prototypically non-canonical ligands, receptors and mediators such as Wnt4, Wnt5a, Wnt7a, Fz2, Ryk or atypical PKC have been observed in the transgenic mouse model of ALS, as we previously discussed in detail. Thus, it is very likely that non-canonical Wnt signaling pathways might be also dysregulated in this neuropathological condition. All the above-mentioned studies and Wnt alterations described in ALS experimental models are summarized in Table 1.
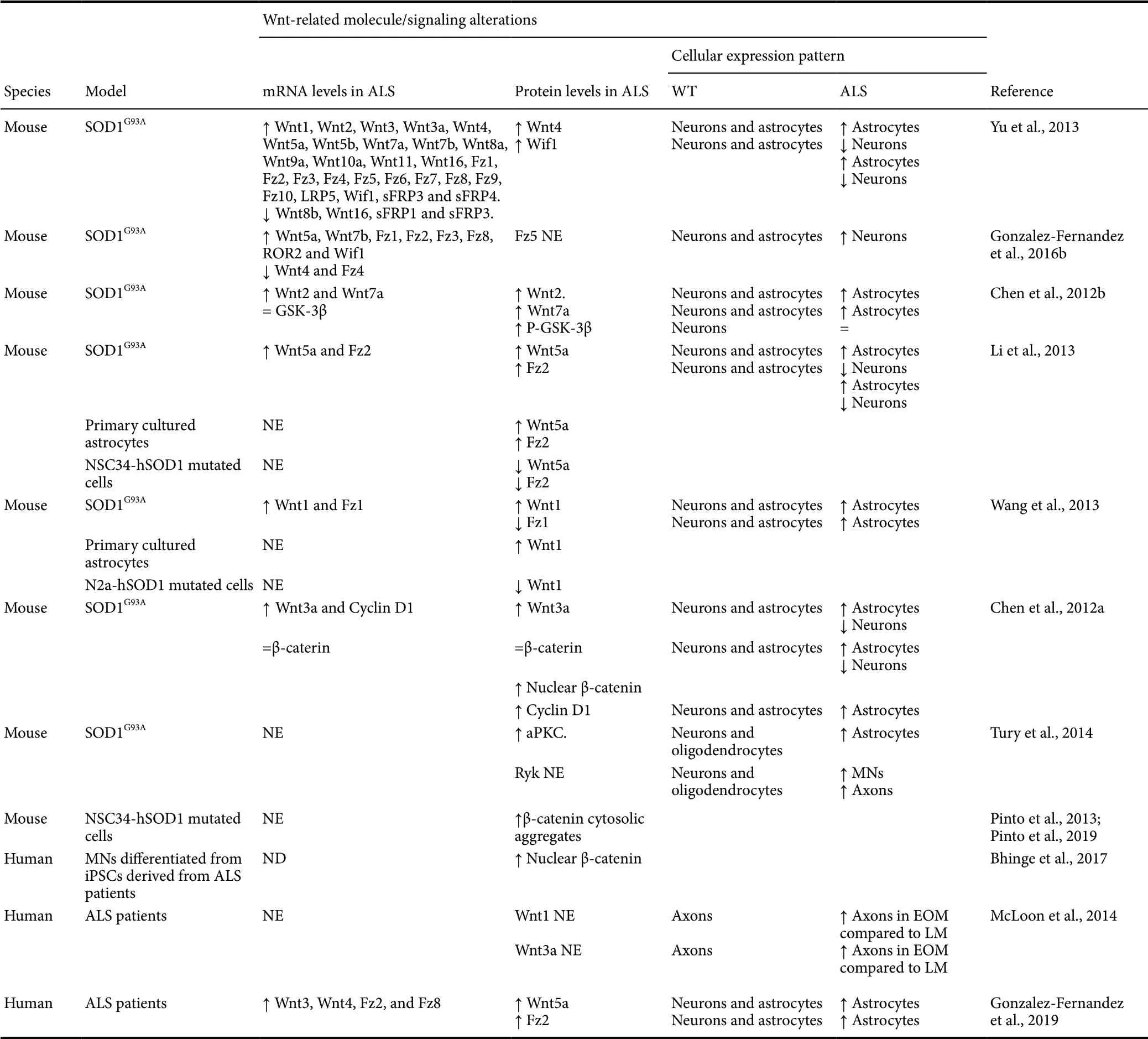
Table 1 Summary of the studies and main findings described in the literature for Wnt-related molecules alterations in ALS
Amyotrophic Lateral Sclerosis-Related Wnt Alterations in Humans
Although experimental models of ALS represent an extremely useful tool to improve our understanding of the pathogenetic mechanisms that characterize this neuropathology, most of them have substantial limitations, since they are only able to model particular or incomplete aspects of the disease, probably leading to a lack of clinic translation. Therefore, to assess the potential clinical relevance of the alterations in the expression of Wnt-related molecules that had been found in experimental models of ALS, it is essential to evaluate whether a differential expression of the Wnt family of proteins also occurs in human ALS pathology.
In this context, there was only one previous study where the authors assessed the existence of potential changes in the expression of Wnt-related molecules concomitant with marked functional sparing of extraocular muscles and their MNs in human ALS (McLoon et al., 2014). More specif ically, when they compared the expression of Wnt1, Wnt3a, Wnt5a and Wnt7a ligands in motor nerves, neuromuscular junctions and muscle fibers between extraocular muscles and limb muscles, they found that Wnt1 and Wnt3a showed a differential expression pattern between healthy and ALS individuals, pointing to a possible role of Wnt signaling in the ocular MNs sparing in ALS.
Interestingly, a recent study performed by our group have shed light on the potential occurrence of Wnt signaling alterations in the pathogenesis of human ALS (Gonzalez-Fernandez et al., 2019), increasing the information limited to the extraocular musculature system and their MNs above-mentioned. In this study, we evaluated the differential gene expression pattern of most of the Wnt signaling components, including all ligands, receptors and soluble modulators in the anterior horn of the spinal cord between controls subjects and ALS cases. Two important and novel findings arise from these analyses: i) most of the Wnt family members are constitutively expressed in the adult human spinal cord, and ii) there is a noticeable gene expression dysregulation of different Wnt family members in the pathological ALS spinal cord. Specif ically, the mRNA levels of Wnt3, Wnt4, Fz2, and Fz8, were significantly upregulated in the anterior horn of the spinal cord of ALS cases, although several non-significant increasing trends were also observed for other genes such as Wnt2b, Wnt5a, Fz3, LRP5 or sFRP3. Interestingly, some discrepancies can be found between these observations and those performed in animal models of ALS, which might suggest the potential existence of between-species differences. However, it is important to highlight that the different experimental approaches used might be underlying these apparent discrepancies, since we performed our gene expression analyses in the anterior horn of the spinal cord, which is one of the main affected areas in ALS, whereas ALS transgenic mice studies were carried out in the entire spinal cord. Overall, our results point to a global Wnt signaling dysregulation associated with this pathology similar to that observed in the mice model of ALS. Interestingly, the Wnt-related molecule that suffered the highest upregulation was the Fz2 receptor, which was detected in neurons and astroglial cells in both healthy and ALS tissue samples. However, a marked increase in the amount of Fz2 expressing astrocytes was found in the white matter bordering the grey matter in the ventral horn and, to a lesser extent, in the grey matter. Additionally, it seems to be an inverse correlation between the amount of Fz2+astrocytes observed and the amount of spared neurons in the ventral horns of ALS spinal cords, suggesting the association between the stage of neurodegeneration and the amount of Fz2+astrocytes in the affected regions. Interestingly, these findings substantially resemble those described in the SOD1G93Atransgenic mouse model of ALS both in vivo and in vitro in primary cultured astrocytes (Li et al., 2013). Furthermore, and analogously to what is described in the mouse model, we also observed an increase of Wnt5a protein levels in ALS human spinal cord (even though no significant mRNA changes were found), specif ically in astrocytes located in the same areas where the alterations in the presence of Fz2+astrocytes were detected. However, in contrast to our previously detailed observations performed in mice, we did not detect any increase in neuronal Fz5 immunoreactivity in the spinal cord of human cases of ALS. Indeed, in the ALS spinal cords we observed a reduction in the amount of Fz5+neurons, which might be related to the loss of neurons that take place in this neuropathological condition. This mismatch might be explained by several reasons, including the potential existence of differences between species, the influence of the different experimental methods used and/or the limitations of the ALS transgenic mice model. All the above-mentioned studies and Wnt alterations described in human samples of ALS patients are summarized in Table 1.
Potential Functions of Wnt Signaling in Amyotrophic Lateral Sclerosis
The current knowledge derived from both experimental models and human ALS cases concerning Wnt signaling disturbances in ALS points to a major role of this family of proteins in this neuropathology. Noticeably, most of the described changes, both in the expression of Wnt components as well as in the activation state of its associated signaling pathways, occur in astrocytes and MNs, which are considered key cellular players in the ALS pathology.
On the one hand, it is well known that astrocytes are crucial cells in the CNS, since they regulate the environment by developing supportive and homeostatic functions. However, in neuropathological states astrocytes are activated, and then proliferate and migrate to the affected areas, where they develop a complex and multifaceted response which allows them to exert both beneficial and deleterious functions (Barbeito et al., 2004; Yamanaka and Komine, 2017). Interestingly, a previous report strongly suggested the potential involvement of the canonical Wnt/β-catenin pathway in astroglial proliferation in ALS. More specif ically, the authors observed an increase in the presence of proliferative Cyclin D1+astrocytes in the ventral horns of the ALS transgenic mice, concomitant with an increase in the presence of nuclear β-catenin in this cell type (Chen et al., 2012a). Otherwise, as is currently well known, astroglial cells are critically involved in pathological neuroinflammation (Colombo and Farina, 2016), which is a crucial process in the pathogenesis of ALS and other neurodegenerative diseases (Hooten et al., 2015; Li et al., 2019). In this regard, a previous study performed by our group has shown that both Wnt1 and Wnt5a, which are upregulated in ALS spinal cords, induce a pro-inf lammatory response in non-activated and activated primary astroglial cultures (Gonzalez and Rodriguez, 2017). Moreover, astroglial cells can also modulate pathological neuroinf lammation by interacting with microglial cells. Accordingly, it has been shown that the interaction between astrocytes and microglia, which express a wide range of Wnt receptors and thus are able to respond to a variety of Wnt ligands (Halleskog et al., 2011, 2012; Halleskog and Schulte, 2013; Gonzalez and Rodriguez, 2017), modif ied neuroinf lammation and disease progression in ALS transgenic mice (Yamanaka et al., 2008; Trias et al., 2018). Interestingly, a previous report has shown that astroglial Wnt5a drives a pro-inflammatory transformation in microglial cells by inducing an increase in the expression of a wide range of inf lammatory mediators as well as in the proliferative capacity of this cell type (Halleskog et al., 2012), in consistence with the widely described pro-inf lammatory role of this Wnt ligand (Blumenthal et al., 2006; Schaale et al., 2011). Similarly, it has been shown that Wnt5a, which is robustly upregulated in astroglial cells in ALS spinal cords due to the activation of the pro-inflammatory transcription factor NF-ĸB in this cell type, induced microglial proliferation in the ALS transgenic mice (Ouali Alami et al., 2018).
On the other hand, another major hallmark of ALS pathogenesis is the selective degeneration of MNs (Mancuso and Navarro, 2015). In this sense, there is growing evidence in the literature regarding the role of Wnt signaling in determining the balance between neuronal survival and death in a variety of neurodegenerative and neuropathological conditions (Inestrosa and Toledo, 2008; Libro et al., 2016). Accordingly, GSK-3β inhibition has been shown to reduce MN death in transgenic ALS mice (Koh et al., 2007; Ahn et al., 2012, 2014). Furthermore, data from both in vitro and in vivo studies have pointed to a neuroprotective role of astroglial-derived Wnt1 via neuronal Fz1/β-catenin signaling in response to oxidative stress and inf lammation (L’Episcopo et al., 2011a, b), in agreement with the different studies pointing to the neuroprotective role of the canonical Wnt/β-catenin signaling pathway in a wide range of neuropathological conditions (Alvarez et al., 2004; Inestrosa and Toledo, 2008; Toledo et al., 2008; Mastroiacovo et al., 2009; Libro et al., 2016). Noticeably, Wnt1 was found to be expressed mainly in spinal cord neurons of WT mice, but the expression of Wnt1 was upregulated in astrocytes of ALS mice spinal cord and in derived primary cultured astrocytes, whereas its proteins levels were found downregulated in an in vitro model of MN-like ALS cells (Wang et al., 2013). Moreover, total Fz1 protein levels were significantly downregulated in ALS mice spinal cords, probably due to a decrease in the neuronal expression of this receptor and/or to a reduced presence of Fz1+neurons, since the expression of this prototypic canonical Wnt receptor was observed in neuronal and astroglial cells and the amount of Fz1-expressing astrocytes is evidently higher in the affected areas in this neuropathological condition (Wang et al., 2013). Accordingly, and as previously detailed, the activity of the canonical Wnt/β-catenin signaling pathway in the affected neurons seems to be reduced, since the presence of β-catenin expressing neurons was decreased in the spinal cord of ALS transgenic mice (Chen et al., 2012a), the transcriptional activity of β-catenin was reduced in an in vitro model of mutated hSOD1 expressing MN-like cells (Pinto et al., 2013), and the cytosolic β-catenin aggregate structures were increased in ALS MNs both in vivo and in vitro (Pinto et al., 2019). Taking into account all the previously detailed observations, it is tempting to hypothesize that the increased astroglial Wnt1 expression might represent a compensatory (and failed) cellular mechanism to try to reduce MN death through Wnt/β-catenin signaling activation in this neuropathological condition.
Otherwise, the involvement of non-canonical Wnt signaling pathways in neuronal survival and death processes is less clear, and contradictory findings have been observed. For instance, it has been shown that Wnt5a, through the non-canonical Wnt/Ca2+signaling pathway, increases neuronal survival by preventing cell cycle activation in Aβ42treated cortical neurons both in vivo and in vitro (Zhou et al., 2017). In contrast, other authors have observed that Wnt5a, whose expression is up-regulated in neuron cultures treated by Aβ peptide, contribute to neurotoxicity (Li et al., 2011). Although the reasons underlying these apparent discrepancies are currently unknown, it is feasible that basal or low levels of Wnt5a may have a neuroprotective effect whereas sustained upregulation of this molecule, such as that observed in ALS pathology, probably potentiates neurotoxicity. Remarkably and as previously described, neurons harbor Wnt5a receptors such as Fz2, Fz5 and Ryk (Li et al., 2013; Tury et al., 2014; Gonzalez-Fernandez et al., 2016a, 2019) in the ALS spinal cords, clearly indicating the capacity of this cell type to respond to Wnt5a.
Finally, evidences from animal models of ALS and human cases suggest that MN pathology could begin at the distal axon and nerve terminals and progress in a dying back pattern to the cell body, and that the alteration of axon guidance molecules may underlie, at least in part, the pathogenesis of ALS (Fischer et al., 2004; Schmidt et al., 2009). Interestingly and as previously detailed, the non-canonical receptor Ryk, a well-known axon guidance molecule under both physiological and pathological conditions (Bovolenta et al., 2006), was found to be upregulated in MNs and white matter axons in the ventral lumbar spinal cord of these ALS transgenic mice. In this regard, although the potential role of this Wnt receptor in axonal dysfunction in this neuropathology is currently unknown and should be evaluated in future studies, it is interesting to note that a previous study performed in Drosophila has found that both Wnt5a and Ryk are critically involved in the proper formation of the neuromuscular junction during development (Liebl et al., 2008). In summary, although the potential specif ic functions of the different components of the Wnt family of proteins are still far away from being well understood, all these evidences strongly suggest the potential involvement of Wnt signaling dysregulations in extremely relevant ALS pathophysiology mechanisms, including proliferation and activation of astroglial and microglial cells, neuroinf lammation, axonal dysfunction and the balance between neuronal survival and death (Figure 2). Therefore, the modulation of specif ic components of this family of proteins as well as its associated signaling pathways might be a promising therapeutic approach in the future.
Potential Therapeutic Approaches
Despite all of the previously detailed evidences pointing to a major role of the Wnt family of proteins in ALS, only a small number of experimental studies have been performed to address the therapeutic potential of Wnt components and/or Wnt signaling pathways modulation in this neuropathological condition.

Figure 2 Schematic representation of Wnt-related signaling dysregulations and their hypothetical involvement in ALS neuropathology.
Noticeably, it has been reported that Riluzole, which is the main currently accepted clinical treatment for ALS, is able to activate the canonical Wnt/β-catenin signaling pathway in different cell types including neuronal cells (Biechele et al., 2010). These interesting observations, together with the previously stated neuroprotective role of this signaling pathway, suggest that the activation of the Wnt/β-catenin signaling pathway could be a promising therapeutic approach for the treatment of ALS. Accordingly, lithium, which is a wellknown activator of the canonical Wnt/β-catenin signaling through GSK-3β inhibition (Meffre et al., 2014), has also been evaluated as a treatment for ALS in both the mouse ALS animal model and in patients, showing a notable attenuation of the disease progression (Fornai et al., 2008). Unfortunately, subsequent clinical trials which investigated lithium as a potential treatment for this neuropathological condition did not f ind evident beneficial effects in ALS patients (UKMND-LiCALS Study Group et al., 2013) while, as previously stated, Riluzol only provide modest benefit. However, a major concern of lithium and Riluzole is their non-specificity, since they not only activate the canonical Wnt/β-catenin signaling but also modulate many other signaling pathways (Noh et al., 2000; O’Brien and Klein, 2009; Yip et al., 2009; Tsuchioka et al., 2011; Daniel et al., 2013). As a consequence, different new GSK-3β inhibitors displaying a higher specif icity and efficacy have been subsequently generated, and some of them have been tested in animal models of ALS showing promising results (Koh et al., 2007; Ahn et al., 2012, 2014), although their therapeutic potential have not yet been evaluated in ALS patients. Besides, it should be noted that more specif ic experimental approaches to activate the canonical Wnt/β-catenin signaling pathway, such as the administration of recombinant Wnt1 or blocking antibodies against Dkk1, are currently under pre-clinical assessment as potential therapies for other neurodegenerative diseases (L’Episcopo et al., 2011a; Purro et al., 2012; Wei et al., 2013) and, therefore, are interesting candidates for the treatment of ALS.
On the other hand, non-canonical Wnt signaling modulation might also be considered an attractive target for the development of novel therapies for ALS neuropathology. For instance and based on the above-mentioned existing evidences described in both ALS transgenic mice and humans, Wnt5a signaling inhibition could be a promising approach for ALS treatment in order to reduce neuroinflammation and glial activation and proliferation which, as previously described, greatly determine the progression and outcome of the disease in both animal models and ALS patients (Hooten et al., 2015). Interestingly, different studies have shown that the Wnt5a-derived hexapeptide known as Box5 is able to selectively inhibit the effects of Wnt5a in different experimental conditions (Jenei et al., 2009; Li et al., 2011). More importantly, it has been demonstrated that the blockage of Wnt5a signaling through Box5 significantly reverses pathological neuroinf lammation in vivo (Yuan et al., 2018). In addition, other therapeutic tools could be used in order to inhibit Wnt5a signaling, such as specif ic Wnt5a neutralizing antibodies. Furthermore, since Wnt function directly depends on the presence of a particular receptor, antibodies against specif ic Wnt5a receptors, such as Fz2, Fz5 or Ryk, could also be used.
Finally, multiple factors should be considered regarding the design and use of Wnt signaling modulation treatments. On the one hand, the highly hydrophobic nature of Wnt ligands (Logan and Nusse, 2004) as well as their short halflife may difficult the administration and distribution of these molecules as therapeutic agents. The development of alternative strategies to increase their half-life and deliver them directly and exclusively to the tissue or cell type of interest (via nanoparticles or other kind of carriers) would be a major improvement in the therapeutic Wnt-based field. Interestingly, it has recently been described the generation of water-soluble tetravalent antibodies with a high specific activity and modular design which enables tailored activation of Fz receptors and LRP5/6 co-receptors, and also allow to provide tissue-specif ic localized activity (Tao et al., 2019). These molecules could depict a key tool for both basic and clinical future research. On the other hand, different studies have clearly related Wnt signaling dysregulations with cancer development and worsening (Logan and Nusse, 2004; Ring et al., 2014). However, although the potential oncogenic effects derived from the modulation of the Wnt family of proteins and its associated signaling pathways should be seriously taken into account, the wide available data from phase I/II clinical trials on different neoplastic diseases as well as other pathological states are being promising (Serafino et al., 2017), and they invite us to believe that Wnt-based treatments could represent an interesting opportunity to explore new therapeutic approaches for ALS pathology.
Conclusion
Evidences taken from recent studies performed in experimental animal models and humans are shedding light on the feasible relationship between Wnt components/signaling pathways dysregulations and the ALS neuropathology. These findings may help us to unravel the underlying pathophysiological mechanisms leading to the MN degeneration and fatal outcome observed in ALS. In this sense, Wnt signaling manipulation could offer a new approach for modulating different critical processes of ALS, such as microglial and astroglial activation, neuroinflammation and/or MN survival. However, further studies are needed to clarify the role of Wnt-related molecules in ALS in order to identify novel therapeutic targets that may lead to new and more effective strategies for ALS treatment through modulation of Wnt signaling pathways.
Author contributions:All authors wrote the manuscript and approved the final manuscript.
Conf licts of interest:We declare no conf licts of interest.
Financial support:This work was supported by the Fondo de Investigación Sanitaria (FIS) of Instituto de Salud Carlos III (Grant number PI12/2895; FEDER co-funded).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Recovery of an injured ascending reticular activating system with recovery from a minimally conscious state to normal consciousness in a stroke patient: a diffusion tensor tractography study
- The role of vascularization in nerve regeneration of nerve graft
- Advanced diffusion magnetic resonance imaging in patients with Alzheimer’s and Parkinson’s diseases
- Modulation of autophagy for neuroprotection and functional recovery in traumatic spinal cord injury
- Decoding epigenetic codes: new frontiers in exploring recovery from spinal cord injury
- Insights into platinum-induced peripheral neuropathy-current perspective