
脂联素抑制β11-肾上腺素受体自身抗体诱导的大鼠H9c2心肌细胞自噬流下降*
2020-06-09 02:41侯晓鸿王昌图王晓晖
中国病理生理杂志 2020年5期
孙 聪, 郭 帅, 宁 娜, 侯晓鸿, 王昌图, 原 媛, 王晓晖,, 王 丽△
(山西医科大学基础医学院 1病理学教研室,2形态学实验室,山西太原030001)
心功能不全是多种心脏疾病的终末阶段,是重要的全球公共健康问题,但发病机制尚未完全阐明[1]。有证据表明,心肌细胞死亡是心功能不全发生发展的关键因素[2]。β1-肾上腺素受体自身抗体(β1-adrenergic receptor antibody,β1-AA)可以诱导心肌细胞死亡[3]。自身抗体是指针对人体自身抗原产生的抗体[4],而β1-AA是针对β1-肾上腺素受体细胞外第2环(the second extracellular loop ofβ1-adrenergic receptor,β1-AR-ECII)产生的抗体[3]。约 40%~60%心功能不全患者血清中均可检测到β1-AA[5],其与心肌细胞膜表面β1-AR-ECII结合发挥类激动剂样作用,引起心肌细胞持续性损伤,导致心肌细胞死亡[3],提示β1-AA诱导的心肌细胞死亡在心功能不全的发生发展中起重要作用。
自噬是细胞内重要的稳态调节机制,自噬流降低可引起细胞内有害蛋白的累积,进而导致细胞死亡[6]。我们课题组前期结果表明,心肌自噬流降低是β1-AA诱导心肌细胞死亡的重要原因,采用自噬激动剂上调心肌细胞自噬流可抑制β1-AA诱导的心肌细胞死亡[7]。因此,逆转β1-AA诱导的心肌细胞自噬流下降成为有效抑制心肌细胞死亡的切入点。
脂联素(adiponectin,APN)可与细胞膜表面脂联素受体(adiponectin receptor,AdipoR)结合,使下游AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)磷酸化,进而上调心肌细胞自噬流[8-9]。APN作为脂肪细胞分泌的细胞因子可增加心肌细胞存活率[10]。然而APN对于β1-AA诱导的心肌细胞死亡及自噬流下降是否具有抑制作用,目前尚无文献报道。因此,本实验拟通过体外研究,观察APN是否可以抑制β1-AA诱导的大鼠心肌细胞自噬流下降,并利用抑制剂干预AMPK磷酸化水平,从分子水平探讨APN调控自噬的可能机制,为β1-AA诱导的心肌细胞自噬流下降的调控提供实验基础。
材料和方法
1 动物和细胞
实验动物:清洁级8周龄雄性Sprague-Dawley(SD)大鼠,体重180~200 g,山西医科大学动物中心[许可证号:SCXK(晋)2015-0001]提供。实验细胞:H9c2大鼠心肌细胞(中国科学院上海细胞库)。
2 主要试剂
β1-AR-ECII抗原肽段(吉尔生化上海有限公司);完全弗氏佐剂和不完全弗氏佐剂(Sigma-Aldrich);重组鼠 APN球状结构域 gAcrp30(PeproTech);LC3B和P62抗体(Abcam);AMPK和p-AMPK抗体(Cell Signaling Technology);Compound C(Selleck);GAPDH抗体和辣根过氧化物酶标记的小鼠IgG(中杉金桥生物公司);Cell Counting Kit-8(CCK-8;日本同仁化学研究所);SYBR Green试剂盒(TaKaRa)。
3 主要方法
3.1 实验分组 将H9c2细胞分为对照(control)组(1μmol/L阴性IgG处理细胞24 h)、β1-AA组(1μmol/Lβ1-AA处理细胞 24 h)、APN+β1-AA组(10μg/L APN预处理细胞1 h后加入1μmol/Lβ1-AA处理24 h)、APN组(10μg/L APN处理细胞25 h)、Compound C+APN+β1-AA组(20μmol/L Compound C预处理细胞30 min后加入10μg/L APN预处理1 h,最后加入1μmol/Lβ1-AA处理细胞24 h)和β1-AA+Compound C组(20μmol/L Compound C预处理细胞30 min后加入1μmol/Lβ1-AA作用24 h)。
3.2 主动免疫大鼠 选取12只8周龄雄性SD大鼠按随机数字表法分为免疫(β1-AR-ECII)组和control组,每组6只。将β1-AR-ECII抗原肽段用Na2CO3溶液溶解稀释,并和完全弗氏佐剂1∶1乳化混匀后,多点背部皮下注射(0.4μg/g),即首次免疫。随后采用已稀释的β1-AR-ECII抗原肽段与不完全弗氏佐剂1∶1乳化,单点背部皮下注射,每隔2周加强免疫1次,共免疫8周;control组用等量Na2CO3溶液替代抗原溶液,实验方案同免疫组。
3.3 亲和层析法提纯大鼠血清中β1-AA 采用亲和层析法提纯主动免疫大鼠血清中的β1-AA,应用于离体实验研究。从4°C取出层析柱在室温下放置30 min,将三蒸水注入层析柱内部,冲去保存在柱子中的乙醇,用结合缓冲液冲洗柱子后,抽取含有β1-AA的血清滤过柱子并用洗脱缓冲液冲洗柱子,使β1-AA随同洗脱缓冲液滴到EP管中,随后用BCA试剂盒进行浓度检测。
3.4 CCK-8实验 本实验采用CCK-8法检测H9c2细胞活力。将细胞进行消化后,大约2×109/L的密度将细胞接种在96孔培养板,每孔100μL细胞悬液,待细胞贴壁后换液,加入5、10和30μg/L APN预处理1 h,随后加入1μmol/Lβ1-AA作用24 h后,每孔加入10μL CCK-8试剂混匀,放入孵箱中2 h,观察到溶液颜色变为棕黄色时,用酶标仪检测在450 nm处各组吸光度(A)值。根据各组的A值计算各实验组相较于对照组的细胞活力。细胞相对活力(%)=(实验组A值-空白对照组A值)/(阴性对照组A值-空白对照组A值)×100%。
3.5 real-time PCR 本实验采用real-time PCR方法检测beclin-1和LC3B的mRNA水平。将细胞进行消化,接种于6孔板中,细胞贴壁后,进行相应预处理,首先采用500μL的PBS进行冲洗,将PBS倒掉并用移液枪将残余的PBS吸净,然后向每孔细胞中加入500μL的TRIzol试剂进行裂解,提取总RNA,0.5μg总RNA反转录为cDNA,选用SYBR Green试剂盒扩增cDNA产物,确认扩增曲线和溶解曲线。引物序列如下:beclin-1(GenBank ID:NM_001034117.1)的上游引物序列为5′-GAAACTGGACACGAGCTTCAAGA-3′,下 游 引 物 序 列 为 5′-ACCATCCTGGCGAGTTTCAATA-3′;LC3B(GenBank ID: NM_022867.2)的 上 游 引 物 序 列 为 5′-AGCTCTGAAGGCAACAGCAACA-3′,下游引物序列为 5′-GCTCCATGCAGGTAGCAGGAA-3′;GAPDH(Gen-Bank ID:NM_017008.3)的上游引物序列为 5′-GGCACAGTCAAGGCTGAGAATG-3′,下游引物序列为 5′-ATGGTGGTGAAGACGCCAGTA-3′。 选 用GAPDH作为内参照。结果采用2-ΔΔCt法进行计算。
3.6 Western blot 本实验采用Western blot方法检测LC3B、P62、AMPK和p-AMPK的蛋白表达水平。将细胞进行消化,接种于6孔板中,待细胞贴壁,进行相应预处理后,向每孔细胞中加入50μL RIPA裂解液、0.5μL PMSF和0.5μL磷酸酶抑制剂。冰上裂解1 h,收取细胞样品,离心后取上清,进行BCA法蛋白浓度检测。选择上样量为40μg,体系为10μL,进行SDS-PAGE分离后,用PVDF膜转膜。再与相应的Ⅰ抗进行反应,4°C过夜,用TBST进行洗膜,与相应的Ⅱ抗在室温下进行反应,用TBST洗膜后,将膜置于全自动曝光仪内,ECL化学发光后,曝光拍摄条带,ImageJ软件分析灰度值,将GAPDH作为内参照,计算不同蛋白的相对表达量。
4 统计学处理
实验数据以均数±标准误(mean±SEM)表示,采用SPSS 16.0软件对数据进行统计分析。两组间比较采用独立样本t检验,多组间比较采用单因素方差分析,两两比较采用LSD-t检验,以P<0.05为差异有统计学意义。
结 果
1 APN抑制β1-AA诱导的H9c2细胞活力下降
CCK-8结果显示,与control组相比,β1-AA诱导心肌细胞活力显著下降(P<0.05),10μg/L APN可显著逆转β1-AA诱导的心肌细胞活力下降(P<0.05),5 μg/L和30μg/L APN均不能恢复β1-AA诱导的心肌细胞活力下降(P>0.05),见图1。因此本研究选择10μg/L为APN的浓度进行后续实验研究。
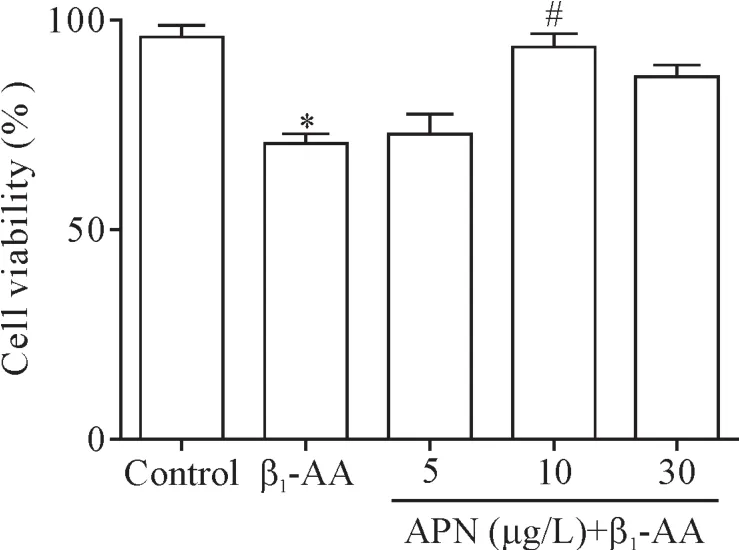
Figure 1.Adiponectin(APN)inhibited the decrease in viability of the H9c2 cells induced byβ1-AA.The H9c2 cells was pretreated with APN(5,10 and 30μg/L)for 1 h before addingβ1-AA(1μmol/L),and CCK-8 assay was performed to measure cell viability.Mean±SEM.n=12.*P<0.05 vs control group;#P<0.05 vsβ1-AA group.图1 APN抑制β1-AA诱导的H9c2细胞活力下降
2 β1-AA诱导H9c2细胞自噬流下降
real-time PCR结果显示,与control组相比,β1-AA诱导beclin-1和LC3B的mRNA水平显著降低(P<0.05),见图2A;Western blot结果显示,与control组相比,β1-AA诱导LC3-II蛋白水平显著降低(P<0.05),P62蛋白水平显著升高(P<0.05),见图2B。
3 APN抑制β1-AA诱导的H9c2细胞自噬流下降
real-time PCR结果显示,与β1-AA组相比,APN显著上调beclin-1和LC3B的mRNA水平(P<0.05),见图3A;Western blot结果显示,与β1-AA组相比,APN预处理可以显著逆转β1-AA诱导的LC3-II蛋白水平下降(P<0.05)及 P62蛋白累积(P<0.05),见 图3B。

Figure 2. β1-AA induced the decrease in autophagic flux of the H9c2 cells.A:the mRNA expression of beclin-1 and LC3B in the H9c2 cells was detected after treatment withβ1-AA(1μmol/L)for 24 h;B:the protein levels of LC3-I,LC3-IIand P62 in the H9c2 cells were measured after treatment with β1-AA(1 μmol/L)for 24 h.Mean±SEM.n=6.*P<0.05 vs control group.图2 β1-AA诱导H9c2细胞自噬流下降

Figure 3. Adiponectin(APN)inhibited the decrease in autophagic flux of the H9c2 cells induced byβ1-AA.A:the mRNA expression of beclin-1 and LC3Bin the H9c2 cells was measured after pretreatment with APN(10μg/L)for 1 h before addingβ1-AA(1μmol/L);B:the protein levels of LC3-I,LC3-II and P62 in the H9c2 cells were detected after pretreatment with APN(10 μg/L)for 1 h before adding β1-AA(1 μmol/L).Mean±SEM.n=6.*P<0.05 vs control group;#P<0.05 vsβ1-AA group.图3 APN抑制β1-AA诱导的H9c2细胞自噬流下降
4 APN抑制β1-AA诱导的H9c2细胞AMPK磷酸化水平下降
Western blot结果显示,与 control组相比,β1-AA显著降低心肌细胞AMPK的磷酸化水平(P<0.05),而与β1-AA组相比,APN预处理可显著改善β1-AA诱导的心肌细胞AMPK磷酸化水平下降(P<0.05),见图4。
5 APN抑制β1-AA诱导的心肌细胞自噬流下降部分依赖AMPK途径

Figure 4.Adiponectin(APN)inhibited the decrease in phosphorylation of AMPK induced byβ1-AA in the H9c2 cells.The protein levels of p-AMPK and AMPK were determined by Western blot.Mean±SEM.n=6.*P<0.05 vs control group;#P<0.05 vsβ1-AA group.图4 APN抑制β1-AA诱导的H9c2细胞AMPK磷酸化水平下降
real-time PCR结果显示,与β1-AA组相比,APN预处理(APN+β1-AA组)可显著逆转β1-AA诱导的beclin-1和LC3B mRNA水平降低(P<0.05);而Compound C预处理(Compound C+β1-AA组)可进一步降低β1-AA诱导的beclin-1和LC3B mRNA水平(P<0.05);当应用Compound C和APN预处理(Compound C+APN+β1-AA组),APN则不能逆转β1-AA诱导的beclin-1和LC3B mRNA水平降低(P<0.05),见图5A。Western blot结果显示,与β1-AA组相比,APN+β1-AA组AMPK磷酸化水平显著升高(P<0.05);而 Compound C+β1-AA 组和 Compound C+APN+β1-AA组AMPK磷酸化水平显著降低(P<0.05),见图5B。进一步检测自噬流,APN+β1-AA组与β1-AA组相比,LC3-II蛋白水平升高,P62蛋白水平下降(P<0.05);Compound C+β1-AA组与β1-AA组相比,LC3-II蛋白水平降低(P<0.05),P62蛋白表达升高(P<0.05);而Compound C+APN+β1-AA组LC3-II和P62蛋白水平均显著下降(P<0.05),即LC3-II的降低无法被APN逆转,但P62的累积仍可被APN抑制,见图5B。
讨 论
心功能不全是全球公共健康问题,患病率和死亡率居高不下[11]。有证据表明,心肌细胞死亡是心功能不全发生发展的关键因素[2]。β1-AA可以与心肌细胞膜表面的β1-AR-ECII结合,导致细胞内环磷酸腺苷累积,影响细胞内Ca2+稳态,进而引起心肌细胞死亡[12]。我们课题组前期研究也表明,β1-AA可导致H9c2心肌细胞活力降低[13]。然而β1-AA如何导致心肌细胞死亡尚不清楚。
研究表明,自噬通过降解细胞内受损、变性及衰老的蛋白质和细胞器实现能量的循环利用,进而维持细胞自身稳态[6]。Miceli等[14]报道,单胺氧化酶通过产生过氧化氢引起氧化应激,诱导H9c2心肌细胞自噬流下降,导致自噬-溶酶体系统清除受损线粒体的能力下降,进而引发细胞内能量代谢紊乱,诱导心肌细胞死亡,提示自噬流降低可能参与H9c2心肌细胞死亡。我们课题组之前的研究结果表明,β1-AA可诱导H9c2细胞自噬流下降,上调心肌自噬流可抑制β1-AA诱导的心肌细胞活力下降,抑制心肌自噬流可导致β1-AA诱导的细胞活力进一步下降[7],提示自噬流降低参与β1-AA诱导的心肌细胞死亡。因此逆转心肌细胞自噬流下降可能有助于抑制β1-AA诱导的心肌细胞死亡。
APN与自噬流密切相关[15]。APN是由脂肪细胞分泌的30 kD的细胞因子[15],具有广泛的心肌细胞保护作用[10]。APN基因敲除小鼠存在严重的心肌自噬流受损,具体表现为自噬小体的生成及清除减少[16],提示APN缺乏可诱导心肌自噬流显著下降。Ahlstrom等[17]报道,在高糖诱导的大鼠骨骼肌细胞中存在自噬流下降,而外源性给予APN可上调骨骼肌细胞自噬流,表明在离体水平APN可抑制骨骼肌细胞自噬流下降。为了证实APN对于β1-AA诱导的心肌细胞自噬流下降是否具有抑制作用,我们首先采用CCK-8法检测各组细胞活力,结果表明,APN可有效逆转β1-AA诱导的心肌细胞活力下降。随后我们进一步采用real-time PCR和Western blot检测各组自噬相关蛋白beclin-1、LC3B和P62的表达水平。beclin-1、LC3B和P62为检测自噬水平的常用指标,其中beclin-1代表自噬的起始,在自噬前体和自噬囊泡的形成过程中起着重要作用[18];LC3B包括LC3-I和LC3-II,LC3-I与脑磷酯结合酯化形成 LC3-II,LC3-II最终定位于成熟的自噬小体膜上,其与自噬小体数量呈正相关[19];P62蛋白是一种选择性自噬底物,当P62蛋白表达增加,表明自噬底物累积,自噬小体清除减少,自噬流降低[19]。本研究结果显示,APN可抑制β1-AA诱导的心肌细胞beclin-1和LC3B mRNA表达下降、LC3-II蛋白水平下降及P62蛋白累积。这提示APN对于β1-AA诱导的心肌细胞自噬流下降具有抑制作用,但具体机制尚不明确。

Figure 5.Adiponectin improved the decreased autophagic flux induced byβ1-AA partially dependent in AMPK signaling.A:the mRNA expressions of beclin-1 and LC3Bwere observed pretreatment with Compound C,adiponectin,or Compound C+adiponectin before addingβ1-AA.B:the protein expressions of P-AMPK,AMPK,LC3 I,LC3 IIand P62 were detected pretreatment with Compound C,adiponectin,or Compound C+adiponectin before adding β1-AA.Means±SEM.n=6.*P<0.05 vs control group;#P<0.05 vsβ1-AA group.图5 APN改善β1-AA诱导的心肌细胞自噬流下降部分依赖AMPK途径
研究表明,APN可通过AMPK途径上调自噬流[20]。AMPK是细胞能量感知和信号调控的重要激酶,对自噬流起着正向调节作用[21]。APN通过与细胞膜表面AdipoR结合,降低细胞内ATP含量,诱导AMPK磷酸化,活化下游Unc-51样自噬激活激酶1(Unc-51 like autophagy activating kinase 1,ULK1)激酶复合物;ULK1是自噬起始的重要激酶复合物,其通过磷酸化beclin-1,进而启动自噬;由此可见,APN可通过AMPK-ULK1途径上调细胞自噬流[20-22]。本研究结果表明,β1-AA可诱导H9c2心肌细胞AMPK磷酸化水平下降,而APN可逆转这一现象。为了进一步证明APN是否通过上调AMPK磷酸化而抑制β1-AA诱导的自噬流下降,我们采用AMPK的抑制剂Compound C抑制AMPK的磷酸化及AMPK介导的自噬激活作用[23]。本研究结果显示,Compound C预处理后,APN无法逆转β1-AA诱导的心肌细胞AMPK磷酸化水平下降,并且自噬流检测结果表明APN也无法逆转β1-AA诱导的LC3B和beclin-1 mRNA及LC3-II蛋白水平的降低,提示APN抑制β1-AA诱导的自噬小体生成降低依赖AMPK途径。同时我们观察到无论是否抑制AMPK的磷酸化作用,APN均可抑制P62蛋白的累积,即可增加自噬小体的清除,提示APN抑制β1-AA诱导的自噬小体清除降低不依赖AMPK途径。上述具体机制尚需进一步探讨。
综上所述,本研究采用β1-AR-ECII主动免疫大鼠血清中提纯的β1-AA作用于H9c2心肌细胞,并外源性给予APN,证实APN可抑制β1-AA诱导的心肌细胞死亡及自噬流降低;同时观察到APN可逆转β1-AA诱导的心肌细胞AMPK磷酸化水平降低;采用Compound C抑制AMPK磷酸化作用后,APN不能抑制β1-AA诱导的自噬小体生成降低,但仍可抑制β1-AA诱导的自噬小体清除降低,提示APN抑制β1-AA诱导的心肌细胞自噬流下降部分依赖AMPK途径。
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国临床解剖学杂志(2022年1期)2022-11-15
检验医学与临床(2022年12期)2022-06-27
中国人兽共患病学报(2022年5期)2022-06-08
世界科学技术-中医药现代化(2022年2期)2022-05-25
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
世界科学技术-中医药现代化(2021年7期)2021-11-04
科学咨询(2020年10期)2020-04-01
科学咨询(2020年1期)2020-02-11
