
二甲双胍激活腺苷酸活化蛋白激酶抑制心肌缺氧再灌注损伤介导的NOD 样受体蛋白3炎症体活化的研究
2020-09-27 09:23杨柳余鹏邹芳蔡霞黄艳婷施星时曼莹张勤江艳娟赖晓阳
中国循环杂志 2020年9期
杨柳,余鹏,邹芳,蔡霞,黄艳婷,施星,时曼莹,张勤,江艳娟,赖晓阳
炎症在心肌缺血再灌注(I/R)损伤中发挥着重要作用[1-2]。NOD 样受体蛋白3(NLRP3)炎性体作为免疫炎症反应的参与者,与心血管疾病关系密切。已有研究证实,NLRP3 炎性体通过多种途径参与心肌缺血再灌注损伤、心肌病、心律失常及冠心病等疾病的发生、发展[3]。细胞凋亡、坏死或炎症均可导致心肌细胞内线粒体三磷酸腺苷(ATP)水平的显著下降,进而激活腺苷酸活化蛋白激酶(AMPK)[4]。AMPK在细胞能量代谢和信号通路激活等过程中发挥关键作用,并且在多种药物抵抗心肌I/R损伤中发挥重要作用[5]。大量基础研究发现,二甲双胍预处理或后处理通过激活AMPK及其介导下游信号通路有效减轻心肌I/R损伤[6]。二甲双胍是否可以通过激活AMPK抑制NLRP3炎症体对心肌发挥保护作用,尚不清楚。本研究拟采用二甲双胍后处理心肌离体缺氧再灌注(H/R)损伤的大鼠模型模拟I/R损伤,探讨二甲双胍介导AMPK激活在抵抗心肌H/R损伤时对细胞内NLRP3炎症体作用及机制。
1 材料与方法
实验分组与处理:选择健康雄性清洁级SD大鼠,体重180~230 g,由南昌大学医学院实验动物中心提供。取健康大鼠64只,进行麻醉,开胸直接取心脏,将心脏悬挂在Langendoff灌注装置上建立离体心肌缺氧再灌注模型。随机分为4组:持续灌注组、缺氧再灌注(H/R)组、二甲双胍后处理组(MET组)、二甲双胍后处理+AMPK抑制剂(compound C,CC)组(MET+CC组),每组16只。除持续灌注组持续灌流3 h外,其余组均先平衡30 min,然后停止灌流30 min,建立离体心肌缺血模型,再持续灌注2 h。MET组及MET+CC组进行后处理:MET组与MET+CC组分别在停止灌流后将二甲双胍(50 µM)及二甲双胍联合复合物C(10 µM)混入K-H液灌注15 min,后继续予以K-H液灌注105 min。
Langendorff离体心肌IR模型制备:根据本课题组之前的研究配置K-H液[7]:NaCl 118.0 mmol/L/、KCl 4.8 mmol/L、KH2PO41.2 mmol/L、NaHCO325.0 mmol/L、MgSO41.2 mmol/L、CaCl22.5 mmol/L和葡萄糖11.0 mmol/L。将PH值调至7.35~7.45,并预先通入95%O2和5%CO2的混合气体30 min,使K-H液与气体充分混合,保持循环温度为37℃。在大鼠腹腔内注射戊巴比妥钠(50 mg/kg)麻醉及股静脉注入肝素(500 U/kg)肝素化后,迅速开胸取心脏,将其置于4℃ K-H液中,排尽残血后悬挂于Langendoff灌注装置上。K-H缓冲液在恒压80 mmHg(1 mmHg=0.133 kPa)、恒温37℃下持续灌注,并吹入95%O2和5%CO2混合气体,通过左心房切口向左心室插入一个与传感器相连的乳胶球囊,向球囊内注水,维持压力于0~10 mmHg。并且利用生物信号采集处理系统(U/4C501H Med Lab,中国上海)采集缺血前即刻(T0)与再灌注30 min(T1)、60 min(T2)、90 min(T3)和120 min(T4)各时间点的血流动力学指标:心率(HR)、左心室峰压(LVSP)和左心室舒张末期压力(LVEDP)。
心肌梗死面积测定:再灌注结束时,立即迅速取下Langendoff灌注装置上的心脏。冷冻(-80 ℃)5 min后,用心脏切割器制成5~6块2 mm厚的组织,置于1% TTC(批号:T8877,Sigma公司,美国)均匀染色。避光、恒温下孵育20 min并于磷酸缓冲盐溶液(PBS,成分为 NaCl、Na2HPO4、KCl、KH2PO4,pH=7.35)冲洗后,10%甲醛液(福尔马林)固定24 h。梗死区及非梗死区分别被染为灰白色和砖红色。用Alpha View凝胶图像软件评估左心室梗死体积/左心肌总体积(即心肌梗死面积)。
心肌组织原位缺口末端标记法(TUNEL)染色测定心肌凋亡:在4℃条件下,多聚甲醛(4%)固定大鼠心肌组织,乙醇脱水、石蜡包埋后进行切片。先用PBS冲洗2遍,按照试剂盒操作步骤,加TUNEL混合溶液,37℃避光孵育1 h。续用4,6-联脒-2-苯基吲哚(DAPI)对比染色细胞核,室温下孵育10 min,PBS冲洗2遍。荧光显微镜下观察并拍照,TUNEL染色呈绿色荧光为凋亡细胞,细胞核呈蓝色荧光,心肌细胞凋亡率=凋亡心肌细胞数/心肌细胞总数×100%。
心脏组织病理染色:于再灌注结束时,立即切取1 mm3左心室置于4% 多聚甲醛固定,制备冷冻切片。将大鼠心脏组织用4%多聚甲醛溶液浸泡24 h以上,乙醇脱水、石蜡包埋处理后切片制成厚4~6 µm的切片,进行常规苏木素-伊红(HE)染色和马松(MASON)染色,于200倍镜下观察心脏形态学变化。
心肌组织酶含量测定:大鼠心脏缺氧再灌注2 h后,取左心室组织,生理盐水制成10%组织匀浆液,离心(4℃、3 000 r/min)15 min后取上清液,利用全自动生化分析仪(Olympus公司,日本)测定乳酸脱氢酶(LDH)、肌酸激酶同工酶(CK-MB)含量。
免疫蛋白印记(Western blot)法测定蛋白表达:于再灌注末迅速取下H/R损伤模型制备成功的左心室心肌组织,剪碎,TBST溶液洗涤2次,细胞裂解液裂解细胞,低温下匀浆混匀(1 2000 r/min),离心10 min(4℃,r=210 mm),取上清液,二喹啉甲酸(BCA)法测定蛋白含量后,制成蛋白样品。取蛋白样品30 µg,与PBS缓冲液充分混匀,浴热5 min。经SDS-PAGE电泳、转印、封闭后,分别加入一抗磷酸化AMPK(1:1 000)(批号:sc-33437,CST公司,美国)、一抗AMPK(1:1 000)(批号:sc-8312,CST公司,美国)、一抗NLRP3(1:1 000)(批号:ab214185,abcam公司,美国)、一抗活化含半胱氨酸的天冬氨酸蛋白水解酶-1(caspase-1)(1:1 000)(批号:89332,CST公司,美国)、一抗白细胞介素(IL)-1β(1:1 000)(批号:12242,CST公司,美国)、和内参抗-甘油醛-3-磷酸脱氢酶(GAPDH;1:2 000;批号:sc-492,CST公司,美国),一抗4℃孵育过夜,二抗室温孵育2h。经洗涤、显影、灰度扫描后,用Image J软件分析条带灰度值,以目的条带灰度值与GAPDH灰度值的比值反映蛋白的表达。
统计学处理:采用Graph Pad Prism 7.00软件进行分析。计量资料以均数±标准差(±s) 表示,组间比较采用单因素方差分析。P<0.05为差异有统计学意义。
2 结果
2.1 血流动力学指标比较(表1)
各组间T0时LVSP、HR和LVEDP差异均无统计学意义(P均>0.05);与T0比较,除持续灌注组外,其他各组T1~T4时LVSP和HR降低,LVEDP升高(P均<0.05);在T1~T4四个时间点,与持续灌注组比较,其他各组LVSP和HR均降低,LVEDP升高(P均<0.05);与H/R组比较,MET组LVSP和HR升高,LVEDP降低(P均< 0.05);与MET组比较,增加AMPK抑制剂的MET+CC组LVSP、HR降低,LVEDP升高(P均< 0.05)。
表1 各组心肌缺氧再灌注期间的血流动力学指标(±s)
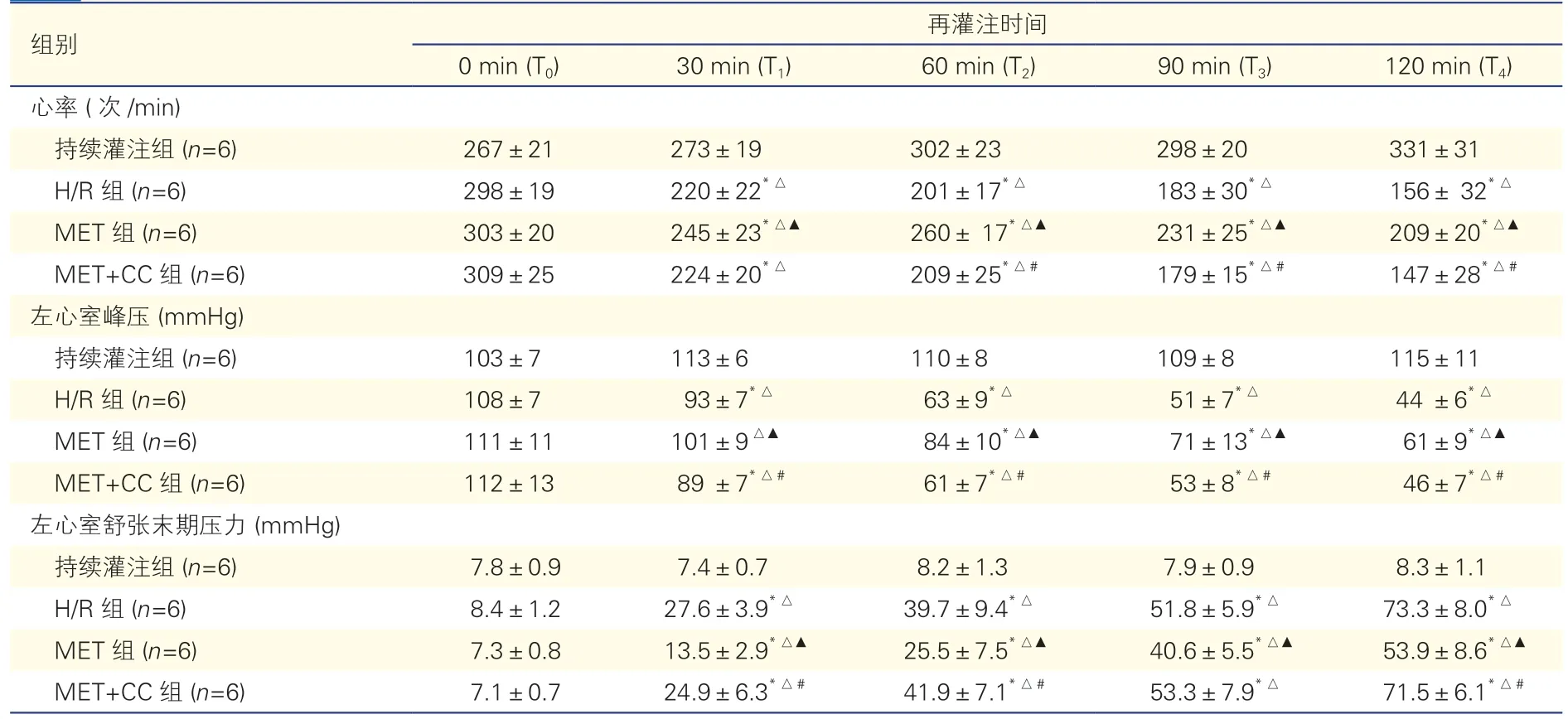
表1 各组心肌缺氧再灌注期间的血流动力学指标(±s)
注:AMPK:腺苷酸活化蛋白激酶;H/R组:缺氧再灌注组;MET组:二甲双胍后处理组;MET+CC组:二甲双胍后处理+AMPK抑制剂组。与T0时比较 *P<0.05;与持续灌注组比较△P<0.05;与H/R组比较▲P<0.05;与MET组比较#P<0.05。1 mmHg=0.133 kPa
2.2 心肌梗死面积和LDH、CK-MB水平比较(表2)
与持续灌注组比较,H/R组心肌梗死面积增加(P<0.05);与H/R组比较,MET组心肌梗死面积减少(P<0.05);与MET组比较,MET+CC组心肌梗死面积增加(P<0.05);与持续灌注组比较,H/R组LDH和CK-MB水平均显著增加(P均<0.05);与H/R组比较,MET组LDH与CK-MB水平均显著下降(P均<0.05);与MET组比较,MET+CC组LDH与CK-MB水平显著升高(P均<0.05)。
2.3 病理组织染色与TUNEL染色(图1)
HE染色显示持续灌注组心肌组织结构清楚,心肌细胞排列整齐,未见心肌细胞坏死、炎症细胞浸润及纤维增生;H/R组心肌细胞排列紊乱,心肌细胞膨大,其间可见小血管扩张、充血,大量炎症细胞浸润及纤维增生;与H/R组比较,MET组心肌组织结构较清楚,心肌损伤较轻,心肌间质少量炎症细胞以及心肌间胶原纤维堆积,其间小血管扩张、充血较轻;而MET+CC组心肌组织损害情况则介于IR组与MET组之间(图1A)。利用MASON染色检测各组心肌间质胶原沉积,光镜下观察可见,持续灌注组心肌间质仅见少量胶原沉积;H/R组胶原沉积较持续灌注组显著增多;MET组与H/R组比较,心肌间胶原纤维堆积明显减少;MET+CC组心肌组织胶原阳性染色较MET组明显增多(图1B)。
表2 各组心肌梗死面积和LDH、CK-MB水平(±s,n=6)

表2 各组心肌梗死面积和LDH、CK-MB水平(±s,n=6)
注:LDH:乳酸脱氢酶;CK-MB:肌酸激酶同工酶;AMPK:腺苷酸活化蛋白激酶。H/R组:缺氧再灌注组;MET组:二甲双胍后处理组;MET+CC组:二甲双胍后处理+AMPK抑制剂组。与持续灌注组比较*P<0.05;与H/R组比较△P<0.05;与MET组比较▲P<0.05

图1 再灌注末各组心肌组织病理改变和心肌组织坏死程度(×200)
2.4 各组大鼠心肌组织TUNEL染色及凋亡率(图2)
TUNEL染色结果显示,与持续灌注组相比,IR组心肌组织内绿色荧光明显增多,心肌细胞凋亡增加(P<0.05);而MET组较IR组绿色荧光显著减少,心肌细胞凋亡指数明显降低(P<0.05)。MET+CC组较MET组心肌组织内绿色荧光增多,心肌细胞凋亡率升高(P<0.05)。
2.5 各组大鼠心肌组织内AMPK/NLRP3通路相关蛋白及IL-1β水平变化(图3)
与持续灌注组比较,H/R组心肌内磷酸化AMPK、NLRP3、活化caspase-1和IL-1β的表达均增加(P均<0.05);而进一步与持续灌注组和H/R组相比,MET组心肌内磷酸化AMPK/AMPK比值明显升高(P<0.05),而NLPR3、活化caspase-1和IL-1β表达水平明显降低(P均<0.05);与MET组相比,MET+CC组心肌内磷酸化AMPK/AMPK比值显著降低(P<0.05),NLPR3、活化caspase-1和IL-1β表达水平均升高(P均 < 0.05)。

图2 再灌注末各组心肌组织TUNEL染色及心肌细胞凋亡率比较
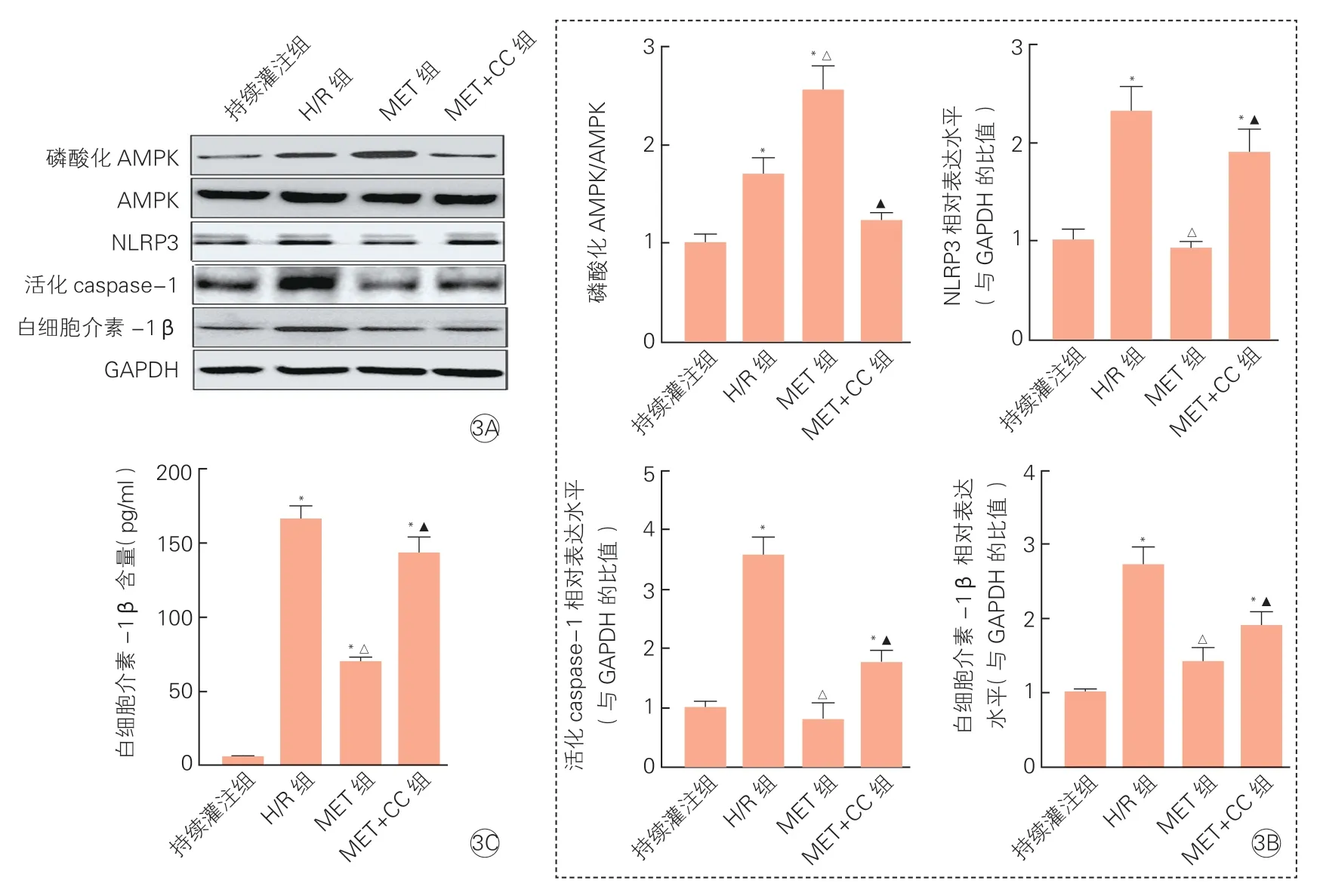
图3 各组大鼠心肌组织内磷酸化AMPK、NLRP3、活化caspase-1和白细胞介素-1β蛋白表达水平(±s,n=3)和白细胞介素-1β含量测定(±s,n=6)
3 讨论
本实验通过观察Langendorff模型制备成功的离体大鼠心脏血流动力学指标,并测定心肌梗死面积、心肌损伤相关酶LDH及CK-MB的含量等来监测各组大鼠心肌H/R损伤。研究表明,MET组较H/R组血流动力学指标明显改善、心肌梗死面积减小、LDH和CK-MB含量下降,结果证实二甲双胍可有效降低心肌H/R损伤。此外,再给予AMPK抑制剂后,相比MET组,MET+CC组大鼠心脏的血流动力学指标显著恶化,心肌梗死面积增大,LDH和CK-MB含量升高。进一步利用HE染色观察心肌组织细胞,结果显示二甲双胍后处理降低了心肌炎症细胞及纤维细胞浸润数量,减轻心肌细胞水肿及间质充血程度;MASON染色结果提示心肌缺氧再灌注损伤导致心肌组织间质胶原沉积明显增多,二甲双胍后处理可显著减少心肌组织胶原沉积。TUNEL染色结果H/R组心肌凋亡率明显升高,二甲双胍后处理可显著降低心肌凋亡率,再次验证了二甲双胍对心肌H/R损伤的保护作用,提示二甲双胍减少H/R损伤所致组织坏死及心肌细胞凋亡,而该保护作用可被AMPK抑制剂所消除。与最新研究结果一致,本研究发现二甲双胍通过激活AMPK有效减少H/R损伤。
AMPK是一种AMP依赖的蛋白激酶,几乎存在于哺乳动物所有组织细胞中,其不仅在调节能量和物质代谢起着重要作用,而且参与细胞增殖、凋亡、自噬,炎症等多种生物学功能。药物或缺血预处理等方式通过激活的AMPK及下游信号通路,发挥调节能量代谢、减轻心肌细胞的炎症反应、抑制心肌凋亡和调节自噬等多种机制对心肌H/R损伤发挥保护作用[8-10]。本研究采用二甲双胍后处理心肌缺氧再灌注损伤的大鼠模型,通过测定AMPK蛋白水平,发现二甲双胍激活AMPK有效抑制H/R损伤诱导心肌细胞炎症反应及凋亡。已有大量研究证实,炎症反应能够加重心肌缺血再灌注或缺氧复氧损伤的组织坏死及凋亡,NLRP3炎症体在其中发挥重要作用[11]。
作为炎症体NOD样受体(NLR)家族中的代表之一,NLRP3是目前研究最为广泛的炎症复合蛋白体。NLRP3可被多种类型的分子、细菌及病毒所激活。NLRP3活化后其N-末端热蛋白结构域(PYD)与含有半胱天冬酶募集结构域的凋亡相关斑点状蛋白质(ACS)结合,同时聚集caspase-1形成NLRP3炎症体[12]。在自身催化作用下caspase-1前体被激活,活化caspase-1可剪切IL-1β和IL-18的前体,随后活化的IL-1β及IL-18等作为主要效应因子,在炎症中发挥重要作用,参与了心肌H/R发生发展[13-15]。Ma等[16]发现中成药清开灵注射液可通过激活AMPK,抑制NLRP3炎症体活化减轻炎症反应,在脑缺血再灌注损伤发挥有益作用。那么二甲双胍对心肌H/R的保护作用是否通过激活AMPK抑制NLRP3炎症体活化?通过Western blot法测定磷酸化AMPK、NLPR3、活 化caspase -1、IL-1β等蛋白的表达,我们发现二甲双胍激活AMPK抑制NLRP3表达并显著减少活化caspase-1和IL-1β水平,最终抑制NLRP3介导的心肌细胞炎症损伤。CC是一种有效的、可逆的、选择性AMPK抑制剂,通过变构调节和抑制AMPK在Thr172的磷酸化激活而发挥效应。我们通过运用CC抑制AMPK激活后,研究发现二甲双胍心肌保护作用显著废除,此外心肌细胞坏死程度显著增加。进一步给予AMPK抑制剂CC处理后的NLPR3表达上调,并且活化caspase-1和IL-1β蛋白表达升高。图1B所示CC抑制AMPK后心肌IR诱导炎症反应导致心肌细胞坏死显著增加,因此我们的结果提示AMPK/NLPR3炎症小体信号通路在二甲双胍抗心肌H/R诱导炎症反应中发挥关键作用。
综上所述,二甲双胍后处理可通过激活AMPK抑制NRPL3炎症体活化进而减少IL-1β等炎症因子水平,减少心肌梗死面积,改善心肌组织病理学损伤,抑制心肌细胞凋亡,最终发挥其对离体大鼠心肌H/R损伤的保护作用。因此,激活AMPK及其调控的NRPL3炎症体活化可能为临床防治缺血性心脏病提供潜在的治疗靶点,并为二甲双胍心肌保护的相关研究提供新的思路。
利益冲突:所有作者均声明不存在利益冲突
猜你喜欢
汽车实用技术(2022年15期)2022-08-19
材料与冶金学报(2022年2期)2022-08-10
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
科学与财富(2021年33期)2021-05-10
今日农业(2020年20期)2020-12-15
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
科学咨询(2020年10期)2020-04-01
科学咨询(2020年1期)2020-02-11
