
营养性肥胖大鼠模型肠道菌群的微生物多样性分析
2020-12-09 03:41李宏睿张德琴金晓蕾王大文辛鹏飞何进程麻富斌周彩云孙晓玮张建刚
中国比较医学杂志 2020年11期
李宏睿,张德琴,金晓蕾,唐 骁,王大文,高 雪,辛鹏飞,何进程, 麻富斌,周彩云,孙晓玮*,张建刚*
(1.兰州大学基础医学院病理学研究所,兰州 730000; 2.山丹县人民医院内科,甘肃 山丹 734100)
随着生活水平的提高,超重和肥胖的人数迅速增加,肥胖症已成为健康的主要危险因素之一。能量摄入过多是肥胖发生的主要原因,作为能量物质最终进入机体内环境(吸收)的门户和屏障,小肠具有重要的研究价值[1]。越来越多的证据表明肠道菌群参与营养性肥胖的发生[2-5]。人的肠道内有大约10万亿个细菌,大多分布于结肠,由于样本主要从粪便中获得,目前大多数研究实际为结肠菌群研究[6-8],然而结肠粪菌是否能够反映小肠的菌群特征,反映与吸收功能及营养性肥胖的确切关系,尚缺乏研究。结肠和小肠菌群差异是营养性肥胖发生的肠道菌群机制研究要解决的关键科学问题[9]。
本文基于前期建立的营养性肥胖大鼠模型,通过16S rRNA测序,对高脂膳食性大鼠模型回肠和结肠内菌群的微生物多样性进行分析,以明确小肠和结肠菌群的分布特点及其与营养性肥胖发生的关系。
1 材料和方法
1.1 实验动物
清洁级雄性Sprague-Dawley(SD)大鼠43只,4~5周龄,体重(52.32±8.48) g,购于兰州大学实验动物中心 [SCXK(甘)2018-0002],。按照实验要求,为模拟自然状态,所有动物在兰州大学病理学研究所动物实验室普通环境下单笼饲养[SYXK(甘)2018-0002],自由进食水。动物实验符合国家实验动物福利相关规定,并获得兰州大学医学伦理委员会批准(jcyxy20190302),实验过程严格按照《实验动物管理与使用指南》进行,严格遵守动物使用的“3R”原则,实验过程给予大鼠以人道主义关怀。
1.2 主要试剂与仪器
高脂饲料按照猪油(lard)∶蛋黄粉(egg yolk powder)∶基础饲料(chow powder)∶蔗糖(sugar)=1∶1∶1.5∶0.4配制[10],能量5.58 kcal/g。猪油从市场购买,新鲜炼制;蛋黄粉购自北京金健力蛋粉厂(蛋白质31.6%,脂肪55.1%,碳水化合物5.3%,巴氏灭菌);SPF级基础饲料(能量3.16 kcal/g)购自北京科澳协力有限公司,60Co 灭菌;蔗糖从市场购买。NucleoSpin 96 Soi DNA提取试剂盒,德国Macherey Nagel公司; KOD FX Neo DNA聚合酶,日本Toyobo公司;Phusion DNA聚合酶,美国NEB公司;DNA纯化柱,美国Omega Biotek公司;Monarch DNA胶回收试剂盒,美国NEB公司。ZK-26/100型真空泵,杭州米欧仪器有限公司;SynergyHTX型酶标仪,美国Gene公司;高速离心机,美国Eppendorf公司;9902型PCR仪,美国Allen Bradley公司。
1.3 实验方法
1.3.1 实验分组及营养性肥胖大鼠模型建立
动物按体重随机分为普通饲料组(chow group,n=10),高脂饲料组(high fat diet, HFD group,n=33),适应性喂养10 d后,进行混合喂养[10],高脂饲料组同时饲喂高脂饲料和普通饲料,普通组单纯饲喂普通饲料,建立营养性肥胖模型。混合喂养60 d,分别以各组前15%为肥胖倾向(>对照组平均体重20%, obese prone, HFD-OP,n=5),以后15%为肥胖抵抗(obesity resistance, HFD-OR,n=5),再以普通饲料继续喂养60 d,去除HFD食物本身诱导对肠道菌群的影响[11-13]。
1.3.2 肠道内容物收集
动物麻醉后皮肤消毒,无菌条件下开腹,门静脉采血处死动物,提取回肠及结肠,分离内容物,并立即于冰上分装至2 mL无菌EP管中,每管样品量0.5~2.0 g,液氮速冻,-80℃保存备检。
1.3.3 微生物多样性分析
利用Illumina测序平台(百迈客生物科技公司,北京),采用Illumina HiSeq 2500双末端测序(paired-end)检测微生物多样性,测序长度350 bp~450 bp。
PCR扩增及测序文库构建:提取菌群总DNA,根据保守区设计细菌16S rRNA基因V3+V4区引物,引物序列如下:338F:5′-ACTCCTACGGGAGG CAGCA-3′;806R:5′-GGACTACHVGGGTWTCTAAT-3′。引物末端加barcode,对目标区域PCR扩增,扩增程序如下:95℃ 5 min→95℃ 30 s→50℃ 30 s→72℃ 40 s 25 cycles→72℃ 7 min→4℃。扩增产物经纯化、定量和均一化,形成测序文库。
测序:PCR扩增产物质检后制备flow cell芯片,Illumina Hiseq 2500双端测序,得到原始图像数据文件,经碱基识别(base calling)分析转化为原始测序序列(sequenced reads)。
数据预处理:根据PE reads之间的overlap关系,将Illumina测序得到的双端序列数据拼接(FLASH, v.1.2.11, JHU Center for Computational Biology)[14],将拼接得到的序列进行质量过滤(Trimmomatic, v.0.33, USADELLAB.org)[15],去除嵌合体(UCHIME, v.4.2, USEARCH 11)[16],成一条序列tags。
OTUs(operational taxonomic units)分析:对测序数据进行质量评估,使用Usearch软件(v.10.0)[17]对tags在97%的相似度水平下进行聚类,获得OTU,一个OTU代表一个物种。使用Mothur软件(version v.1.30, http://mothur.org/),对各个样品的alpha多样性指数进行评估,使用QIIME软件(v2.2, SCIKIT-BIO, http://qiime.org/)进行beta多样性分析,比较不同样品在物种多样性方面存在的相似程度。
物种注释及分类学分析:将OTU的代表序列与Silva微生物参考数据库(v. 128, http://www.arb-silva.de)[18]进行比对,得到每个OTU对应的物种分类信息,应用RDP Classifier(v2.2, QIIME)[19]对OTU进行分类学注释。进而在门、纲、目、科、属、种(phylum, class, order, family, genus, species)各水平统计各样品群落组成,利用QIIME软件生成不同分类水平上的物种丰度表,R语言绘制群落结构图。
1.4 统计学方法
2 结果
2.1 营养性肥胖模型
建模60 d,普通饲料组体重为(326.51±29.29) g,高脂饲料组体重为(355.99±36.33) g(P<0.05)。高脂饲料组15%个体体重(HFD-OP)> 对照组平均体重20%(5/33),与普通饲料组及相应15%低体重(HFD-OR)(5/33)相比,差异具有统计学意义(P< 0.05),HFD-OR与普通饲料组相比,差异无统计学意义(P>0.05)(表1)。
24 h初始进食量普通饲料组为(9.23±2.01) g,高脂饲料组为(9.10±1.71) g,差异无统计学意义,P>0.05。建模期间,普通饲料组进食量为(980.00±53.43) g(普通饲料),高脂饲料组进行混合喂养,其中高脂饲料的进食量为(630.70±90.61) g,普通饲料的进食量为(337.88±82.34) g,高脂饲料与普通饲料的比值为(2.02±0.72)∶1,提示实验动物更偏好高脂饲料。
HFD-OP大鼠在高脂饲料、普通饲料的进食量以及对食物的偏好与HFD-OR大鼠无显著性差异(P>0.05)(表1)。
2.2 低能量膳食对营养性肥胖的影响
经普通饲料继续喂养60 d,OP大鼠的体重仍然显著高于OR与普通饲料组,其肾周及睾周脂肪质量也显著高于其它组,P<0.05,见表2。
表1 普通饲料组与高脂饲料组大鼠体重及摄食情况Table 1 Body weight and food intake of rats in chow group and HFD group
2.3 肠道菌群多样性分析
提取肠内容物总DNA,样品质量符合测序要求,利用Illumina测序平台对样品DNA进行扩增、测序,得到2489179个PE reads,经质量过滤后共得到2208772个clean tags。各样品GC含量为(54.21±0.97)%,Q30达到(93.96±1.61)%。测序长度在406~420 bp之间。
OTU分析:OTU数反映菌群的多样性。使用Usearch软件对tags在97%的相似度水平下进行聚类、获得OTU。经过60 d的饲料均一化干预,高脂饲料组回肠OTU数与普通饲料组无明显差异,但结肠OTU数显著降低,表明高脂膳食引起大鼠结肠微生物丰度下降,见表3。
不同体重回肠与结肠OTU数无显著性差异,见表4。
Alpha多样性分析回肠、结肠菌群的OTU,获得ace、chao1、simpson、shannon指数,多元多因素方差分析显示,体重、饲料、部位因素之间无交互作用(P>0.05,数据未提供),饲料与体重对alpha多样性指标无明显影响(P>0.05,数据未提供),但回肠与结肠间alpha多样性差异具有统计学意义,回肠菌群的chao1、ace指数均显著高于结肠,表明回肠菌群中细菌数量(丰度)远高于结肠,而多样性低于结肠(simpson、shannon)(P<0.05)(表5)。Venn图显示,除普通饲料回肠外,其余组内各样品间OTU具有较大的重叠性,图1。
2.4 回肠与结肠菌群beta多样性分析
应用非加权Binary_Jaccard距离算法比较不同样品在物种多样性方面存在的相似程度,UPGMA聚类树图显示样品间菌群结构相似,但回肠与结肠样品间的相似度较低(图2)。
表2 低能量膳食对营养性肥胖大鼠体重及内脏脂肪的影响Table 2 Effects of low energy diet on body weight and visceral fat of nutritional obese rats
表3 普通饲料组及高脂饲料组回肠、结肠菌群OTU数比较Table 3 Comparison of OTUs in ileum and colon of chow group and HFD group
表4 不同体重大鼠回肠、结肠菌群OTU数比较Table 4 Comparison of OTUs in ileum and colon of rats with different weights

注:除普通饲料回肠,各组个体间共有的OTU数显著高于个体特有的OTU数。图1 回肠、结肠细菌OTU的Venn图Note. Except for chow-ileum, the number of Common OTUs in each group was significantly higher than that of individual’s specific OTUs.Figure 1 Venn diagram of OTU in ileum and colon
表5 回肠、结肠菌群alpha多样性指数比较
进一步采用非度量多维标度(nonmetric multidimensional scaling, NMDS)、主坐标分析(principal coordinate analysis, PCoA)和主成分分析(principal component analysis, PCA),对肠道微生物群落结构的变化进行分析,如图3所示,结肠样本间菌群组成显示出明显的相似性,而回肠与结肠以及回肠样本间相似度较低。
2.5 物种注释及分类学分析
将OTU的代表序列与微生物参考数据库Silva进行比对,得到每个OTU对应的物种分类信息。结果显示,在属水平上,回肠与结肠优势菌(丰度排名top 10)存在比较大的差异,回肠内的主要优势菌为Romboutsia、Turicibacter、Rothia、Lachnospiraceae、Streptococcus、Candidatus_Arthromitus、Helicobacter、Lactobacillus等,而结肠的主要优势菌为Lachnospiraceae、Treponema、Akkermansia、Candidatus_Saccharimonas、Ruminococcaceae、Romboutsia等,两者优势菌的重叠程度较低,见图4、图5。
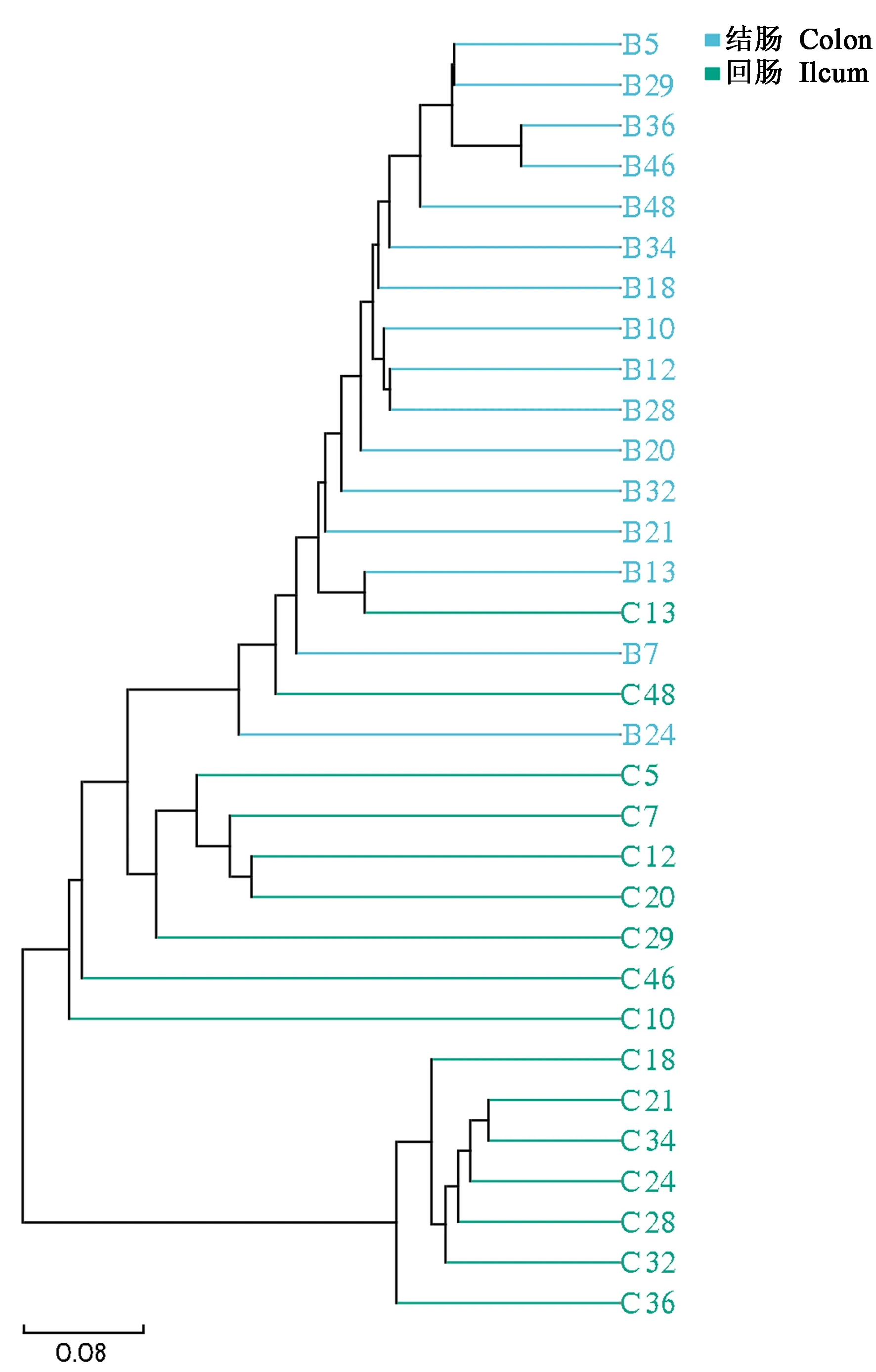
注:B:结肠;C:回肠。图2 UPGMA聚类树图显示回肠、结肠细菌 结构特征Note. B, Colon. C, Ileum.Figure 2 UPGMA cluster tree showed the structural characteristics of ileum and colon microbiota
HFD大鼠回肠Turicibacter低于普通饲料组,而结肠内的Treponema、Candidatus_saccharimonas低于普通饲料组,证明高脂膳食对回肠和结肠菌群的影响不同,并且这种影响具有持久性。
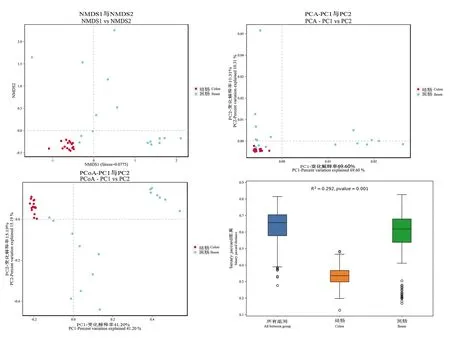
图3 NMDS、PCA、PCoA分析法显示回肠、结肠细菌结构特征Figure 3 NMDS, PCA and PCoA analyses showed the structural characteristics of ileum and colon microbiota

图4 回肠内细菌相对丰度和物种注释(属水平)Figure 4 Relative abundance and bacteria annotation in ileum (genus level)

图5 结肠内菌群相对丰度和物种注释(属水平)Figure 5 Relative abundance and bacteria annotation in colon (genus level)

注:B:结肠;C:回肠。图6 HFD-OP回肠、结肠内细菌物种丰度聚类热图(属水平)Note. B, Colon. C, Ileum.Figure 6 The cluster heat map of bacterial species abundance in the ileum and colon of HFD-OP rats (genus level)
回肠是营养物质吸收的主要部位,HFD-OR大鼠回肠内Rothia菌丰度明显增加,而Romboutsia菌丰度降低,提示Rothia菌和Romboutsia菌可能参与回肠吸收的调节作用。此外,HFD-OR结肠内Ruminococcaceae_UCG-005显著增加。
聚类分析热图显示HFD-OP大鼠回肠与结肠内菌群类型的差异,HFD-OP大鼠回肠内Lachnospiraceae、Candidatus_Arthromitus、Helicobacter、Lactobacillus较其它处理组丰度低,而Turicibacter、Romboutsia、Streptococcus、Rothia的丰度较高。与其它各组相比,结肠内菌群除与其它各组相同的优势菌群外,Prevotella_9、Prevotellaceae_Ga6A1_group丰度较高,而Candidatus_Saccharimonas、Lactobacillus、uncultured_bacterium_f_Ruminococcaceae丰度较低,图6。
3 讨论
营养性肥胖是一种严重威胁人类健康的全球公共卫生问题,肠道作为能量物质最终进入机体内环境的门户和屏障,其内大量定居的菌群被认为在肥胖的发生中发挥着重要作用[20]。肠道菌群作为机体重要的共生生命,与食物成分及消化道的功能相互协同,形成一个微生态系统,在消化、营养、免疫等方面发挥重要生理功能,若其构成改变也会导致心、脑等器官发生多种严重疾病[21-23]。哺乳动物肠道微生物数量存在纵向变化,例如在人的消化道内,细菌数量呈指数级增长[24],胃、小肠近端、回肠、结肠中每毫升内容物的细菌数量分别为102~103、103~105、108、1011。结肠是机体最大的食物残渣分解场所,长期以来,肠道菌群的研究主要集中在结肠,那么结肠粪菌能否代表小肠菌群?本研究表明,尽管回肠与结肠之间菌落的OTU数差异不明显,并且多数菌群存在重叠(Venn图),但alpha多样性分析显示回肠菌群的样品内物种丰度显著高于结肠(chao1、ace),而多样性低于结肠(simpson、shannon);beta多样性分析显示结肠样本间菌群组成具有明显的相似性,而回肠与结肠以及回肠样本间相似度较低;经注释分析,两者的优势菌群存在明显的不同,并且重叠程度较低;聚类分析热图也显示回肠与结肠内的菌群物种丰度分布不同,表明结肠内的粪菌并不能代表回肠内的菌群结构。因此,在肠道菌群研究中必须注意这一问题,这与Leite等[25]对人、Grond等[26]对滨鸟、Su等[27]对蒙古马的研究结果相一致。
小肠是营养物质吸收的主要场所,研究结论显示,肠道菌群通过多种机制参与肥胖的发生,包括LPS诱导的肠渗漏和炎症[28],慢性低度内毒素[29]、肠源性肽激素[30]、脂肪组织活性成分等的调节[31],包括GPCR、AMPK信号途径影响能量平衡和饱食感,以及短链脂肪酸影响脂类和能量物质代谢[32]等,但多数研究是基于结肠粪菌探讨与肥胖的关系,还不足以揭示小肠菌群调节吸收促进营养性肥胖发生的机制。我们的研究发现,回肠内的主要优势菌为Romboutsia、Turicibacter、Rothia、Lachnospiraceae、Streptococcus、Candidatus_Arthromitus、Helicobacter、Lactobacillus等,而结肠的主要优势菌为Lachnospiraceae、Treponema、Akkermansia、Candidatus_Saccharimonas、Ruminococcaceae、Romboutsia等,提示回肠与结肠菌群存在功能的差异,小肠菌群和粪菌可能具有不同的功能属性。
OTU是一个相对稳定的指标,不容易受到营养程度的影响,但本研究表明,高脂膳食可能对结肠的OTU数存在比较持久的影响,物种注释显示高脂膳食对回肠、结肠内的优势菌也具有持久的影响。我们发现,Turicibacter、Rothia、Romboutsia、Streptococcus可能参与回肠吸收的调节,在营养性肥胖的发生中发挥一定的作用,对于这一结果,还需要更多的动物实验研究支持。
肠道菌群研究是一个非常复杂的领域,一方面由于肠道微生物组成在个体间具有很大的差异,而且不同背景(饮食习惯和种族)的参与者构成及用于分析细菌的不同方法在很大程度上都会影响研究结果的统一;另一方面则因为疾病动物模型在肠道微生物群组成、发酵过程、进食习惯等方面存在差异[33]。本文在营养性肥胖大鼠模型的基础上,研究肠道菌群与肥胖发生的关系,进一步证实结肠与回肠具有不同的菌群特征,对于探寻人类肠道菌群促进营养性肥胖发生的机制具有较好的借鉴价值。
综上所述,本研究通过对高脂膳食性营养性肥胖大鼠回肠和结肠菌群进行16S rRNA基因测序分析,证实回肠与结肠具有不同的肠道菌群,结肠菌群并不能代表回肠的菌群特征,肠道的部位是进行肠道菌群研究必须要考虑的因素;Turicibacter菌、Rothia菌、Romboutsia菌、Streptococcus菌可能是参与肥胖发生的关键回肠固有菌群。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
特别健康·下半月(2020年11期)2020-12-15
中华养生保健(2020年5期)2020-11-16
初中生学习指导·提升版(2020年3期)2020-09-10
中国生殖健康(2018年1期)2018-11-06
健康大视野(2018年5期)2018-07-16
中华胰腺病杂志(2018年3期)2018-01-12
今日健康(2016年7期)2017-04-12
医食参考(2017年12期)2017-04-01
中国动物保健(2015年4期)2015-10-21
