
SIAP:一种蛋白质复合物识别分布式算法
2021-01-21 12:58邓超刘桂霞孙立岩王荣全
哈尔滨工程大学学报 2020年11期
邓超, 刘桂霞, 孙立岩, 王荣全
(1.吉林大学 软件学院,吉林 长春 130012; 2.吉林大学 计算机科学与技术学院,吉林 长春 130012)
近邻传播聚类算法(affinity propagation clustering AP算法)[1]是2007年由Frey等提出并发表在《Science》上的聚类算法。与其他的 聚类算法相比,AP算法有较高的精度,在应用方面AP算法有较少的约束条件,但在适应不同形式的数据方面相对不足,肖宇等[2]提出SAP(semi-supervised Affinity propagation)算法,利用数据中的先验信息,调整相似性矩阵,在先验性信息较多的情况下,算法适应性较好。AP算法特点在于,参考度的值与聚类结果相关,因此本文融合ECC[3]与CD[4]方法对AP算法进行改进,动态设置参考度,不再简单的设置为所有数据中位数的值。
我们进入了大数据时代,在大数据中挖掘出有用的信息,已经成为科技发展的主流趋势[5]。单一的串行模式耗时长、效率低。为了解决这个问题,一些大数据计算平台随之产生。其中基于内存计算的Spark[6]分布式平台倍受关注。它的生态圈十分丰富,包括特定场景下的计算库(Streaming、SQL、MLlib、Graphx)。此外其数据保存在内存中,减少了多次计算中磁盘的I/O开销。AP算法聚类的过程中,需要在矩阵之间不断地进行迭代替换,算法运算时间较长,面对海量数据,单机串行实现AP算法,不能满足实验计算需求。为了改进对处理海量数据的适应能力。本文提出基于Spark并行运算改进的AP算法SIAP(spark based on improved AP),提高算法的运行速率。
蛋白质复合物是由2个以上功能相关且相互作用连接在一起的蛋白质组成[7]。目前,蛋白质复合物的识别手段分为生物实验和计算方法两大类,生物实验方法持续周期长、实验成本高,而计算方法可以更好的弥补生物实验技术的不足。通过不断的发展,出现很多蛋白质复合物识别算法,MCODE算法检测网络中蛋白质链接相对密集的簇为蛋白质复合物[8],PCP采用FS加权的方法构造有权图,然后在加权网络中通过寻找稠密子图预测蛋白质复合物[9]。本文应用AP算法识别蛋白质复合物。首先,PPI数据形式多样[10],本文对数据进行评分处理,并应用Spark平台对AP算法进行并行化处理,并给出加速比图示。最后分别在黑腹果蝇(dm)和人类(hs)2个真实PPI数据集上对本文改进的AP算法与原始的AP算法、clusterone[11]、OH-PIN[12]、IPCA[13]等算法进行比较,最后引入F-Measure[14]评价指标对结果进行评估,并给出实验效果图。
1 基于ECC与CD改进的AP算法
1.1 AP算法
AP算法的基本思想基于数据点,在数据点之间进行消息的传递,通过迭代运算自动识别聚类中心。AP的计算过程是通过不断更新吸引度矩阵和归属度矩阵实现的。
吸引度r(i,k)中,i为数据对象,k表示作为候选代表点的数据对象,r(i,k)则代表从点i发送到候选聚类中心k的数值消息,反映的是k点是否适合作为i点的聚类中心[15]。吸引度的定义为:
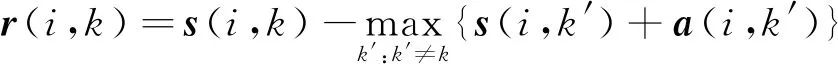
(1)
式中:s(i,k)表示i点与k点之间的相似度;a(i,k)代表归属度,表示从候选中心k发送到i的数值消息,反映i点是否选择k作为聚类中心[15]。归属度的定义为:

(2)
在算法的运算过程中,产生的聚类结果可能不稳定,加入震荡系数,用于调节算法的稳定性:
rt+1(i,k)=λrt(i,k)+(1-λ)rt+1(i,k)
(3)
at+1(i,k)=λat(i,k)+(1-λ)at+1(i,k)
(4)
式中:t和t+1分别代表上一次和本次的迭代结果;λ为震荡系数。
基于式(3)、(4),AP算法的运算过程如下:
1)输入数据生成相似度矩阵,并设置参考度,在原始的算法中一般设为数据中位数的值。
2)输入所需的最大迭代次数和震荡变量λ的值,构造初始为零矩阵的吸引度与归属度矩阵,并进行矩阵的迭代,更新数据。
3)确定聚类中心,以r(k,k)+a(k,k)>0为准则,输出聚类中心以及每个点所属的类别,算法结束。
1.2 基于ECC与CD改进的AP算法
实验中应用的数据是无权的PPI数据,为了达到更好的实验结果,将PPI数据生成的网络中的边标记权重[16],ECC和CD是2种很有效的加权方法[3-4]:
(5)
(6)
式中Nu和Nv分别为蛋白质u和v的邻居数量。
本文将2种方法结合,使加权的精度更高。以此构造相似度矩阵。权重公式定义为:
W(u,v)=βECC(u,v)+(1-β)CD(u,v)
(7)
在构造相似度矩阵时,要设定参考度的值,传统的AP算法将对角线设为统一的定值,本文应用权重结果,动态分配对角线的值,使邻接边多的并且具有更高权重值的数据点,赋值越大,加大成为聚类中心的几率。参考度为:
i,k,n∈m,s(i,k)>0
(8)
式中:s(i,k)为i行中大于0的值;n为当前行s(i,k)的个数;ave为所有边权重的均值。
改进的AP算法的运算步骤如下:
1)根据式(7)、(8),对输入的数据进行打分赋值,生成相似度矩阵,并得出参考度的值。
2)输入最大迭代次数maxIter和震荡变量λ的值,更新迭代吸引度矩阵和归属度矩阵。
3)以r(k,k)+a(k,k)>0为标准,输出聚类中心以及每个点所属的类别,算法结束。
2 基于Spark的改进AP算法
Spark是一个高速运算级别的,具有强大功能的开源式计算引擎。有着Hadoop的MapReduce[17]中的优点,并扩展了MapReduce的计算模式,Spark的分布式计算更高效。Spark整体架构基于4大部分Spark Streaming、Spark SQL、Mlib、Graphx,如图1所示。

图1 Spark架构Fig.1 The architecture of Spark
Spark的主要核心是弹性分布式数据集,即RDD[18](resilient distributed datasets),RDD是一个只读的,缓存在内存中的数据集。在并行计算中应用RDD可以减少内外存的读写操作。并且支持map、reduce、collect等数据操作。此外单机模式,应用到大数据中会有很多限制,达不到运算要求。Spark以集群的模式进行部署,Spark子集群结构如图2所示。集群中一共有2类节点,master负责资源管理,Slave则负责计算。

图2 Spark子群结构Fig.2 The subgroup structure of Spark
基于以上理论基础,本文AP算法进行并行化处理。此外在算法并行的过程中内存会受到限制,RDD应用的数据结构存储方式至关重要,本文将list类型与tuple类型相结合,以三元组的形式存储数据,生成最小内存的RDD,存储结构形式如下:
List[tuple(rowid,rowValues)],其中rowid为行号,rowValues为第rowid行的numpy数组。
读取数据,2条数据之间有边连接,则将数据的编号和边的权值存储在List里,2条数据之间没有边连接则不存储。将相似性矩阵s,吸引度矩阵r,归属度矩阵a数据构建RDD的操作如下所示:
s=sc.parallelize(l,NUM_PARTITIONS);s=s.map(lambdax: f0(x,count))
其中f0为数据格式转换函数,读List进行操作,转换成数据矩阵中行的形式,并应用map函数生成一个新的RDD。
a=sc.parallelize(range(count),NUM_PARTITIO-NS);
a=a.map(lambdat:(t,np.zeros(count)))
r=sc.parallelize(range(count),NUM_PARTITIO-NS);
r=r.map(lambdat:(t,np.zeros(count)))
其中NUM_PARTITIONS是并行分配的参数,count为数据点的个数。
基于Spark平台SIAP算法具体执行过程如下所示:
输入:原始的PPI数据集,打分调节变量β,震荡变量λ,最大迭代次数maxIter.
1)初始化:s[count][count]=0,r[count][count]=0,a[count][count]=0
2)根据式(7)计算相似度,为边加权重,并存储到s中
3)根据式(8)更新参考度的值s[i,i]
4)构建s矩阵,r矩阵,a矩阵RDD
5)初始化i=1
6)根据式(3)更新吸引度矩阵r;
根据式(4)更新归属度矩阵a;
7)l=i+1,如果l 8)如果a(i,i)+r(i,i)>0: 则第i个数据为聚类中心,存储在数组centers中 9)数据分类,计算找出a(i,i)+r(i,i)每行所属聚类中心的最大值,将该点分配到此聚类中心。返回RDD所有元素 10)输出结果,算法结束。 本文将SIAP算法应用于蛋白质复合物识别中,以此来验证SIAP聚类的加速效果,并根据F-measure将SIAP算法与AP、Cytoscape[19]软件中的clusterone、OH-PIN、IPCA这4种算法进行对比,实验中分别采用Drosophila melanogaster(dm)[20]数据集与Homo sapiens(hs)[21]数据集作为测试对象。参照集本文使用Vinayagam等的蛋白质复合物数据集[22]。 本文选择Spark2.4.0作为数据计算框架,操作系统为Ubuntu16.04,python版本为3.5.2,Anaconda版本为4.2.0,分布式环境由5台物理机组成,master为主节点,slave1~slave4为worker节点,各节点的配置如表1所示。 在并行计算中采用加速比来验证效果,加速比是指,同一任务在单处理系统和并行处理系统中运行消耗时间的比率。加速比定义为: (9) 表1 实验环境配置参数 实验应用黑腹果蝇(dm)和人类(hs)2个真实数据集分别在2、4、8、16个节点上运行SIAP算法,算法运行时间如图3所示。 图3 算法运行时间Fig.3 The runtime of algorithm 由图3可知,针对2种数据集,随着计算节点数的增加,2种数据集的运行时间均呈现下降趋势,并且接近线性趋势。说明算法有良好的拓展性,适用于处理不同的数据集,未呈现线性的原因是在算法运行过程中,需要将数据分片处理,在不同的节点间,要进行信息交流,数据传递需要消耗一部分时间。为了使对比更加明显,针对2种数据集的算法加速比如图4所示。 图4 算法加速比Fig.4 The acceleration ratio of algorithm 从图4中可以看出,随着计算节点数的增加,加速比的增加趋势形式正相关,但与线性趋势有一定差距,随着节点数的增多,差距略微变大。未呈现线性的原因,同样是由于节点之间的信息传递需要一定的额外时间。通过Spark平台的应用,缩短了单线程运算的时间,充分体现的算法的并行化的速度优势。 为了验证算法的准确率,引入综合评价指标F值(F-Measure)对识别的蛋白质复合物进行评估,准确率(precision)和召回率(recall)是F值的重要参数。准确率是指算法识别的蛋白质复合物中被匹配部分所占比例,召回率是指已知蛋白质复合物中被匹配部分所占比例,具体为: (10) 式中:TP是指算法识别出的蛋白质复合物与已知复合物匹配的数值;FP是指算法识别出的蛋白质复合物总数减去TP数的值;FN表示不能被算法识别出的蛋白质复合物匹配的已知复合物的数值。 F值的定义为: (11) F值是综合正确率与召回率的一种评估指标,反映整体的综合效果。F值越大,代表所得到的聚类结果越好。基于2个真实dm数据集与hs数据集,SIAP算法与原始的AP算法、clusterone、OH-PIN、IPCA这4个聚类算法进行对比,实验结果如图5所示。 图5 算法对比结果Fig.5 The comparison result of algorithm 通过图5分析可知SIAP与未改进得AP算法在两种数据集中,都有良好的聚类效果。clusterone在2种数据集中,结果相差不大,也表明了clusterone聚类算法对数据集的适应能力。针对不同的数据集SIAP算法的精度高于其他聚类算法,证明了SIAP算法的有效性。 1)引入ECC和CD这2种加权方法,对无权的蛋白质相互作用网络数据进行加权处理。 2)设置动态的参考度,将参考度与边的紧密型关联起来。 3)应用Spark平台对算法进行并行化计算,缩短了算法的运行时间,提高了算法的运行速率。 实验中将SIAP与原始的AP算法以及clusterone、OH-PIN、IPCA算法进行对比,实验结果表明,SIAP算法在不同数据集上均有最高的F值,达到了良好的聚类精度证明了算法的有效性。在接下来的研究中,将考虑生物意义,将聚类算法与生物学信息相结合,提出更有效的蛋白质复合物识别方法。3 实验结果与分析
3.1 实验环境
3.2 实验结果




4 结论
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
中学生数理化(高中版.高考数学)(2022年1期)2022-04-26
西华大学学报(自然科学版)(2020年6期)2020-10-15
数学小灵通(1-2年级)(2020年6期)2020-06-24
现代计算机(2018年27期)2018-10-25
安徽化工(2018年4期)2018-09-03
雷达学报(2017年6期)2017-03-26
中学生数理化·八年级数学人教版(2017年2期)2017-03-25
互联网天地(2016年1期)2016-05-04
现代计算机(2016年17期)2016-02-28
