
抗阿尔茨海默病药物盐酸多奈哌齐的合成
2021-05-06 01:04谢宇轩
合成化学 2021年4期
胡 昆, 谢宇轩, 任 杰
(常州大学 制药与生命科学学院,江苏 常州 213164)
阿尔茨海默病(AD)是一种起病隐匿的、进行性发展的神经系统退行性疾病,具有不可逆性和致死性[1-2]。阿尔茨海默病的病理表现主要包括:细胞外β-淀粉样蛋白沉积导致老年斑形成、细胞内过度磷酸化的Tau蛋白造成神经原纤维缠结以及突触丢失等[3-6]。阿尔茨海默病的发病假说有以下几种:年龄[7]、遗传[8]、Aβ沉积及毒性作用[9]、Tau蛋白过度磷酸化[10]、胆碱能功能缺损[11]、兴奋性氨基酸毒性[12]等。随着人口老龄化的加速,老年人口的比例迅速增加,阿尔茨海默病已成为严重的公共卫生和社会问题,给老年人、患者家庭和社会造成了巨大压力。
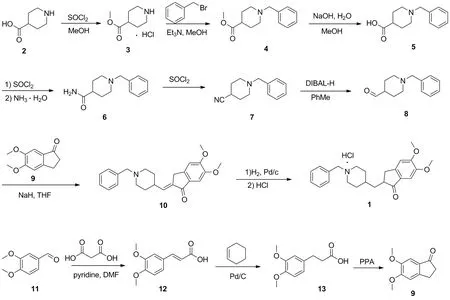
Scheme 1
盐酸多奈哌齐(1)化学名为2,3-二氢-5,6-二甲氧基-2-{[(1-苯甲基)-4-哌啶基]甲基}-1H-茚-1-酮盐酸盐,是第二代可逆性乙酰胆碱酯酶抑制剂,也是FDA批准用于治疗AD的第二种药物[13]。目前,1已经获得世界上30多个国家的认可。与其他治疗阿尔茨海默病的药物相比,1具有显著优点[14-16]:(1)选择性更强,只对中枢神经中的胆碱酯酶有抑制作用;(2)用药量少且给药方便、作用时间长、治疗费用低;(3)无明显毒副作用、不良反应少;(4)明显改善轻度至中度病人的认知能力和临床症状,提高患者的自知力、生活自理能力和自制力。1作为治疗轻度至中度AD的有效药物,其深度开发具有广阔的市场前景。
合成1的方法主要有以下3种:(1)德国拜耳公司[17]提出了以5,6-二甲氧基-1-茚酮和4-吡啶甲醛为主要原料的合成路线,总收率58.5%。该方法经缩合、苄基化和还原等3步反应得到1,虽然步骤较短,但原料价格较为昂贵,且吡啶环的还原条件较为严苛,催化剂价格相对较高,不利于大规模工业化生产。(2)美国辉瑞公司[18]以4-吡啶甲醛为原料,经过缩合、还原、酰化、氯代、Friedel-Crafts反应、缩合、分子内环合、脱保护、苄基化得到多奈哌齐。该合成路线较长,总收率较低,仅19.3%。此外,该路线使用了剧毒物质氯甲酸甲酯,且使用的原料及试剂价格相对昂贵,条件较为严苛,增加了生产成本。(3)Gaonkar等[19]以4-哌啶甲酸甲酯为原料,经烷基化、还原、Swern氧化反应后得到N-苄基-4-哌啶甲醛,再和5,6-二甲氧基-1-茚酮经缩合、还原、成盐反应得到1。 Swern反应条件较为严苛,后处理繁杂,反应过程会产生令人不快的气味,并且雷尼镍不利于大规模工业化生产。
本文设计了一条合成1的新路线。以4-哌啶甲酸(2)为原料,经酯化反应生成4-哌啶甲酸甲酯盐酸盐(3);3经烷基化反应生成N-苄基-4-哌啶甲酸甲酯(4);4水解得到N-苄基-4-哌啶甲酸(5);5经酰化反应生成N-苄基-4-哌啶甲酰胺(6);6脱水得到1-苄基哌啶-4-腈(7);7经还原反应生成N-苄基-4-哌啶甲醛(8);以3,4-二甲氧基苯甲醛(11)为原料,经缩合反应生成3-(3,4-二甲氧基苯基)丙烯酸(12);12经催化转移氢化反应生成3-(3,4-二甲氧基苯基)丙酸(13);13经分子内环化反应生成5,6-二甲氧基-1-茚酮(9);以8和9为原料经过缩合反应生成2-(1-苄基-4-哌啶亚甲基)-5,6-二甲氧基-2,3-二氢-1H-茚酮(10);10经还原反应成盐得1(Scheme 1),总收率56.8%,其结构经1H NMR和13C NMR确证。
1 实验部分
1.1 仪器与试剂
SGW X-4型显微熔点仪;Bruker Avance III 500 MHz型核磁共振仪(DMSO或CDCl3为溶剂,TMS为内标)。
所用试剂均为分析纯。
1.2 合成
(1)3的合成
在100 mL圆底烧瓶中加入4-哌啶甲酸25.00 g(38.71 mmol),加入甲醇(30 mL)将其溶解;滴加氯化亚砜(7.5 mL),滴毕,回流反应2 h(TLC检测)。浓缩反应液得白色固体36.84 g,收率98.4%, m.p.160 ℃;1H NMR(500 MHz, DMSO)δ: 3.62(s, 3H), 3.18(dt,J=12.8 Hz, 3.3 Hz, 2H), 2.94~2.86(m, 2H), 2.73~2.66(m, 1H), 1.97(dd,J=14.3 Hz, 3.8 Hz, 2H), 1.83~1.74(m, 2H);13C NMR(126 MHz, DMSO)δ: 174.11, 52.22, 42.45, 38.05, 24.83。
(2)4的合成
在100 mL圆底烧瓶中加入33.80 g(21.15 mmol),用甲醇(30 mL)将其溶解;依次加入苄溴4.30 g(25.38 mmol)、三乙胺4.70 g(46.53 mmol),回流反应6 h(TLC检测)。减压浓缩反应液,残余物加入水(100 mL),用乙酸乙酯(3×40 mL)萃取,合并有机相,过滤,滤液依次用饱和食盐水(2×150 mL)洗涤,无水硫酸镁干燥,过滤,滤液浓缩得黄色油状液体44.65 g,收率94.2%;1H NMR(500 MHz, CDCl3)δ: 7.35~7.25(m, 5H), 3.69(s, 3H), 3.51(s, 2H), 2.87(d,J=11.7 Hz, 2H), 2.32(ddd,J=11.2 Hz, 7.1 Hz, 4.1 Hz, 1H), 2.07~2.01(m, 2H), 1.92~1.87(m, 2H), 1.84~1.75(m, 2H);13C NMR(126 MHz, CDCl3)δ: 175.67, 138.35, 129.08, 128.20, 126.99, 63.25, 52.91, 51.62, 41.09, 28.30。
(3)5的合成
将43.50 g(15.00 mmol)加入到100 mL圆底烧瓶中,用甲醇(30 mL)溶解;加入10%氢氧化钠溶液(10 mL),回流反应2 h(TLC检测)。浓缩反应液,残余物加入甲醇(50 mL),用37.5%盐酸调至pH≈6,析出白色固体,抽滤,滤液浓缩后得黄色固体53.20 g,收率98.2%, m.p.169.0~170.5 ℃;1H NMR(500 MHz, DMSO)δ: 7.44~7.41(m, 5H), 3.54(s, 2H), 3.13~2.96(m, 2H), 2.50~2.45(m, 1H), 2.23~2.04(m, 4H), 1.96~1.74(m, 2H);13C NMR(126 MHz, DMSO)δ: 178.14, 137.78, 128.56, 128.53, 127.64, 63.16, 50.85, 40.41, 28.26。
(4)6的合成
将53.20 g(14.59 mmol)加入100 mL圆底烧瓶中,加入氯化亚砜(10 mL)溶解;回流反应2 h(TLC检测)。浓缩反应液得黄色油状液体,加入乙腈(5 mL),滴加到冰浴冷却的氨水(30 mL)中,反应0.5 h(TLC检测)。浓缩反应液,析出白色固体,抽滤,滤饼用水(3×50 mL)洗涤,真空干燥得白色固体63.06 g,收率96.3%, m.p.161 ℃;1H NMR(400 MHz, CDCl3)δ: 7.26~7.33(m, 5H), 5.99(s, 1H), 5.63(s, 1H), 3.52(s, 2H), 2.96(d, 2H), 2.19(t, 1H), 2.04(t, 2H), 1.88(d, 2H), 1.73~1.81(m, 2H);13C NMR(100 MHz, CDCl3)δ: 178.03, 138.20, 129.11, 128.22, 127.06, 63.16, 53.05, 42.83, 28.92。
(5)7的合成
将62.80 g(12.83 mmol)加入100 mL圆底烧瓶中,加入氯化亚砜(10 mL),回流反应5 h(TLC检测)。浓缩反应液,残余物加入二氯甲烷(10 mL),用氨水调至pH≈8,分液,水相用二氯甲烷(2×20 mL)萃取,合并有机相,用无水硫酸镁干燥,过滤,滤液浓缩得黄色油状液体72.43 g,收率94.0%;1H NMR(500 MHz, CDCl3)δ: 7.53~7.09(m, 5H), 3.53(s, 2H), 2.69(d, 3H), 2.34(s, 2H), 1.96~1.88(m, 4H);13C NMR(126 MHz, CDCl3)δ: 137.78, 128.56, 128.54, 127.64, 122.75, 63.16, 50.82, 27.18, 26.21。
(6)8的合成
将72.00 g(9.99 mmol)加入100 mL圆底烧瓶中,用甲苯(20 mL)溶解;于0 ℃加入DIBAL-H(1.5 M in Toluene, 8 mL),反应1 h(TLC检测)。加入饱和亚硫酸氢钠溶液(10 mL),用2N氢氧化钠溶液调至pH≈8,用乙酸乙酯(3×30 mL)萃取,合并有机相,依次用饱和食盐水(2×150 mL)洗涤,无水硫酸镁干燥,过滤,滤液浓缩得无色油状液体81.98 g,收率97.5%;1H NMR(500 MHz, CDCl3)δ: 9.67(s, 1H), 7.28~7.34(m, 5H), 3.52(s, 2H), 2.86(d, 2H), 2.24~2.29(m, 1H), 2.15(t, 2H), 1.92(d, 2H), 1.67~1.75(m, 2H);13C NMR(126 MHz, CDCl3)δ: 204.75, 137.78, 128.56, 128.53, 127.64, 63.16, 51.02, 47.08, 26.87。
(7)12的合成
在100 mL圆底烧瓶中加入3,4-二甲氧基苯甲醛115.00 g(30.09 mmol),用DMF(30 mL)将其溶解;加入丙二酸3.70 g(36.11 mmol),吡啶3.6 mL,于90 ℃反应5 h(TLC检测)。倒入冰水(200 mL)中,用浓盐酸调至pH≈2,析出白色固体,抽滤,滤饼真空干燥得白色固体125.20 g,收率82.8%, m.p.179.4~181.1 ℃;1H NMR(500 MHz, DMSO)δ: 7.54(d,J=16.0 Hz, 1H), 7.30(s, 1H), 7.21(d,J=8.0 Hz, 1H), 6.98(d,J=8.0 Hz , 1H), 6.47(d,J=16.0 Hz, 1H), 3.80(d,J=8.0 Hz, 6H);13C NMR(126 MHz, DMSO)δ: 168.37, 151.21, 149.40, 144.62, 127.48, 123.09, 117.12, 111.92, 110.70, 56.01, 55.96。
(8)13的合成
将123.00 g(14.41 mmol)加入100 mL圆底烧瓶中,加入乙醇(40 mL)溶解;加入10%Pd/C 0.15 g,环己烯1.8 mL,回流反应10 h(TLC检测)。抽滤,滤液浓缩得灰白色固体133.00,收率99.0%, m.p.98~99 ℃;1H NMR(500 MHz, DMSO)δ: 6.82~6.84(m, 2H), 6.71~6.73(m, 1H), 3.73(d,J=8.0 Hz, 6H), 2.77(t, 2H), 2.52(t, 2H);13C NMR(126 MHz, DMSO)δ: 174.35, 149.03, 147.53, 133.81, 120.38, 112.65, 112.27, 55.95, 55.82, 36.08, 30.47。
(9)9的合成
将多聚磷酸20 g加入圆底烧瓶中,预热至90 ℃,加入132.00 g(9.51 mmol),搅拌下反应15 min(TLC检测)。倒入冰水中(100 mL),用二氯甲烷(3×40 mL)萃取,合并有机相,依次用饱和碳酸氢钠溶液(2×100 mL)和饱和食盐水(2×100 mL)洗涤,无水硫酸镁干燥,过滤,滤液浓缩得黄色固体91.70 g,收率92.9%, m.p.120 ℃;1H NMR(500 MHz, CDCl3)δ: 7.18(s, 1H), 6.89(s, 1H), 3.96(s, 3H), 3.90(s, 3H), 3.06(t, 2H), 2.68(t, 2H);13C NMR(126 MHz, CDCl3)δ: 205.73, 155.36, 150.41, 149.33, 129.85, 107.46, 104.12, 56.20, 56.06, 36.50, 25.56。
(10)10的合成
将5,6-二甲氧基-1-茚酮91.89 g(9.80 mmol)溶于二氯甲烷(15 mL)中,加入TBAB 0.1 g和10%氢氧化钠溶液7 mL,搅拌30 min;加入82.00 g(9.80 mmol),于室温反应5 h(TLC检测)。旋蒸除溶,粗品用乙酸乙酯重结晶得白色固体103.20 g,收率86. 5%, m.p.175~177 ℃;1H NMR(500 MHz, CDCl3)δ: 7.36~7.26(m, 6H), 6.91(s, 1H), 6.68(d,J=9.7 Hz, 1H), 3.98(s, 3H), 3.94(s, 3H), 3.63~3.58(m, 2H), 3.55(s, 2H), 2.94(d,J=11.8 Hz, 2H), 2.35(dd,J=9.6 Hz, 4.5 Hz, 1H), 2.14~2.00(m, 2H), 1.78~1.55(m, 4H);13C NMR(126 MHz, CDCl3)δ: 192.57, 155.29, 149.49, 144.51, 139.81, 138.16, 135.63, 131.85, 129.21, 128.20, 127.03, 107.24, 105.01, 63.50, 56.25, 56.15, 53.07, 37.27, 31.22, 29.50。
(11)1的合成
将101.00 g(2.65 mmol)加入圆底烧瓶中,用THF(10 mL)溶解;加入10%Pd/C 0.10 g,通入H2反应3 h(TLC检测)。过滤,滤液浓缩,残余物用乙醇(5 mL)溶解;滴加浓盐酸(0.4 mL),滴毕,搅拌1 h;旋蒸除溶,残余物用混合溶剂(乙醇/乙醚=2/1,V/V)重结晶得白色晶体10.90 g,收率81.8%, m.p.211~212℃;1H NMR(500 MHz, CDCl3)δ: 7.35~7.30(m, 4H), 7.26(ddd,J=5.1 Hz, 2.9 Hz, 1.8 Hz, 1H), 7.18(s, 1H), 6.87(s, 1H), 3.98(s, 3H), 3.92(s, 3H), 3.53(s, 2H), 3.25(dd,J=17.6 Hz, 8.1 Hz, 1H), 2.92(t,J=10.7 Hz, 2H), 2.74~2.68(m, 2H), 1.96(dddd,J=22.0 Hz, 9.4 Hz, 7.1 Hz, 3.6 Hz, 3H), 1.78~1.66(m, 2H), 1.51(td,J=7.5 Hz, 6.9 Hz, 2.9 Hz, 1H), 1.41~1.31(m, 3H);13C NMR(126 MHz, CDCl3)δ: 207.82, 155.43, 149.40, 148.76, 138.42, 129.32, 129.24, 128.14, 126.92, 107.35, 104.36, 63.45, 56.22, 53.81, 45.47, 38.72, 34.47, 33.35, 33.03, 31.80。
2 结果与讨论
2.1 合成4的反应条件优化
该反应为N-烷基化反应,影响反应速率及收率的因素主要有烃化剂、原料配比、反应溶剂、反应时间等。主要考察了原料配比[r=n(苄溴)/n(3)]和反应时间对收率的影响。
(1)r
表1为r对4收率的影响。由表1可以看出,当原料配比为 1.2时,收率相对较高,继续增大原料配比,收率趋于稳定。因此反应的最佳原料配比为1.2。
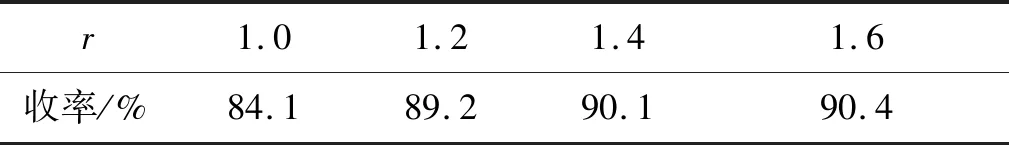
表1 r对4收率的影响Table1 Effect of r on yield of 4
(2) 反应时间
表2为反应时间对4收率的影响。由表2可以看出,当反应时间延长至8 h以后,反应收率趋于稳定,虽然12 h收率最高,但是反应时间较长,增加能耗和不可控因素。综合考虑,最优反应时间为8 h。

表2 反应时间对化合物4收率的影响Table 2 Effect of reaction time on yield of4
2.2 合成7的反应条件优化
该反应为酰胺脱水生成氰基,脱水剂和溶剂对收率有较大影响[20]。为了寻求合成7的最佳反应条件,考察了脱水剂和溶剂对收率的影响。
(1) 脱水剂
表3为脱水剂对7收率的影响。由表3可知,当选择氯化亚砜为脱水剂时,能够以高收率得到产物7。

表3 脱水剂对7收率的影响Table 3 Effect of reaction dehydrating agenton on yield of 7
(2) 反应溶剂
表4为溶剂对7收率的影响。由表4可知,不使用溶剂,能够以高收率得到产物7。

表4 反应溶剂对7收率的影响Table 4 Effect of reaction solvent on yield of 7
2.3 合成9的反应条件优化
该反应为分子内环化反应,分子内自身发生傅克反应,失去一分子水得到五元环。因此要求该反应的催化剂必须同时具有促进酰化和环合的作用。多聚磷酸具有性质温和、环合速度快、收率高、产品纯度好等优点,最终选择作为催化剂。影响环合的主要因素有催化剂用量、反应温度和时间。为了寻求合成9的最佳反应条件,考察了原料比[r=n(多聚磷酸)/n(13)]、反应温度和反应时间对收率的影响。
(1)r
多聚磷酸作为环合剂,其用量对9的收率有较大的影响。以化合物131.0 g为原料,考察原料配比对9收率的影响,所有收率均为3次平行试验的平均值,结果见表5。由表5可知,当原料配比为10时,产品收率相对较高,继续增大原料配比,副反应出现,收率逐渐降低。 因此反应的最佳原料配比为10。

表5 不同条件下9的收率Table 5 Yield of 9 under different conditions
(2) 反应时间和温度
以化合物134.8 mmol为原料,考察不同时间和温度下对9收率的影响,结果见表6。由表6可知,随着反应温度的升高,想要保持较高的收率必须减少反应时间,以减少副产物的生成。通过比较70 ℃、 90 ℃、 110 ℃等若干组反应,发现90 ℃反应15 min的收率最高。

表6 反应时间和温度对9收率的影响Table 6 Effect of reaction time and temperature on yield of 9
以4-哌啶甲酸为原料,经酯化、烷基化、酯水解、酰化、脱水、还原反应得到关键中间体N-苄基-4-哌啶甲醛(8);以3,4-二甲氧基苯甲醛为原料,经缩合、还原、分子内环化反应得到5,6-二甲氧基-1-茚酮(9);8与9经缩合、还原反应后成盐合成了最终产物盐酸多奈哌齐,并对其合成条件进行了优化,总收率为56.8%。与已有合成路线相比,该合成路线虽然相对较长,但具有起始原料廉价易得、操作简便、反应条件温和、后处理简便等优点。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
农业工程学报(2022年5期)2022-06-22
化工时刊(2022年1期)2022-05-25
健康体检与管理(2022年2期)2022-04-15
精神医学杂志(2021年2期)2021-06-23
上海计量测试(2020年1期)2020-03-18
中国医药指南(2019年30期)2019-11-28
中小学实验与装备(2016年1期)2016-04-19
药学研究(2015年12期)2015-12-08
红领巾·探索(2014年8期)2014-10-10
